

NeurIPS 2023 Roundup: Generative Protein Design
Generative protein design took center stage at NeurIPS 2023. In part one of a three part series, we highlight several papers, authors, and takeaways about the intersection of ML and proteins.

Intro
This is Part One of what I expect to be a three part series recapping ML x Bio highlights from NeurIPS 2023. I’ve done my best to tag authors—who should feel free to correct me if I misunderstood anything. I’ve organized these posts topically and aim to publish weekly.
Generative Protein Design
Generative Chemistry
Drug-Target Interaction (DTI) Prediction
Overview
Proteins took center stage across both bio-focused workshops at NeurIPS 2023. Speakers discussed cutting-edge methods for generative protein design, binding site prediction, and protein-protein docking. My main takeaways include:
Optimizing proteins in structure-space is easier than in sequence-space.
Data distributions of protein structure are smoother, making them more tractable to sample from. This came through from several talks on inverse folding. LLMs were notably absent from the bio-workshops compared to diffusion and flow models. As counterargument—structure data is much rarer and more expensive to generate.
There’s still a disconnect between academia and industry.
Many talks focused on binding affinity. Though helpful, industry practitioners seemed more interested in multi-parameter optimization including thermostability, immunogenicity, developability, etc.
Data leakage, overfitting, train-test splits, and experimental validation are key concerns.
Exquisite model performance fell flat unless authors followed up with a meticulous walkthrough of training and test data, benchmarks, and in vitro validation.
Accuracy is great. Scale and throughput matter a lot too.
Many drug discovery applications require scale. As mentioned in our MoML post, inference speed is a deciding factor whether a model is useful in industry.
Let’s get to it.
Protein Design Using Inverse Folding
Inverse folding is a method to predict a protein’s 1D amino acid sequence from its 3D structure. Given the importance of local 3D structure to antibody-antigen binding, many authors used inverse folding as a means to constrain a generative antibody process. More simply, they’d lock in a tight-binding 3D structure and only sample sequences that could satisfy the 3D constraint. My sense is this greatly narrows the search space for multi-parameter property optimization.
In Vitro Validated Antibody Design Against Multiple Therapeutic Antigens Using Generative Inverse Folding
Julian Alverio, an employee of Absci (ABSI), presented on his firm’s continued progress in generative antibody design. Specifically, we learned about the company’s inverse folding framework, IgDesign, and how they wield it to generate antibody candidate designs against known targets. As mentioned earlier, inverse folding is attractive because the data distribution of structure is smaller and smoother (read: easier to optimize) than sequence space.
IgDesign is inspired by models like AbMPNN and and LM-Design, but extends the field by showing in vitro experimental binding results for the designed antibodies. I’ve attached the high-level overview of LM-Design and IgDesign below. My understanding of what’s under the hood is as follows:

IgMPNN makes up the first part of the overall IgDesign workflow. Similar to AbMPNN, IgMPNN is a fine-tuned version of ProteinMPNN optimized for antibodies. Unlike AbMPNN, IgMPNN has access to antigen and antibody framework sequences during training.
IgMPNN’s CDR design function is based on the concept of cross-attention between sequence and structural embeddings as described in LM-Design. Essentially, IgMPNN fuses a structure-aware graph neural network (GNN) with a protein language model (ESM2-3B)—allowing IgMPNN to leverage both spatial and evolutionary information. It’s a bit like consulting two complementary experts before making a final decision.
Finally, the team expressed candidate designs (including single and multiple CDRs) in vitro and tested these against the SAbDab baseline (CDR3) for eight different target antigens, as shown below.

The audience had questions surrounding training data representativeness, multiparametric optimization, and data leakage. One question I have is around the 40% sequence identity training split. When I was at MoML in November, I learned that proteins with ~15% sequence identity can still come from the same family and be structurally similar. Should the training split be lowered, then?
I also heard from diffusion model advocates that because CDR regions are so hypermutable, they aren’t subject to the same evolutionary pressures that other proteins are. Therefore, algorithms reliant on evolutionary information (read: language models) aren’t as effective for antibody CDR design tasks. I’d love to hear the rebuttal!
AntiFold: Improved Antibody Structure Design Using Inverse Folding
Alissa Hummer and Magnus Høie presented their group’s work on AntiFold, another inverse folding model for generative antibody design. I appreciated how the speakers cast inverse folding as a means to constrain sequence space. That is, having a desired final structure allows the model to sample only mutations that don’t alter antibody structure. I imagine this constraint is useful for co-optimizing other salient properties using language models (e.g., stability, expression) more efficiently.
AntiFold is fine-tuned from ESM-IF1 using a combination of experimental (n=2,074) and predicted (n=147,458) antibody structures from SAbDab and OAS, respectively. I’d like to better understand the pitfalls of training on predicted structures. The authors mask ~15% of the protein’s backbone with a bias towards antibody CDR loops as these are much more variable. AntiFold’s training objective is to fill back in masked sequences while maintaining the antibody’s structural identity, as highlighted below.
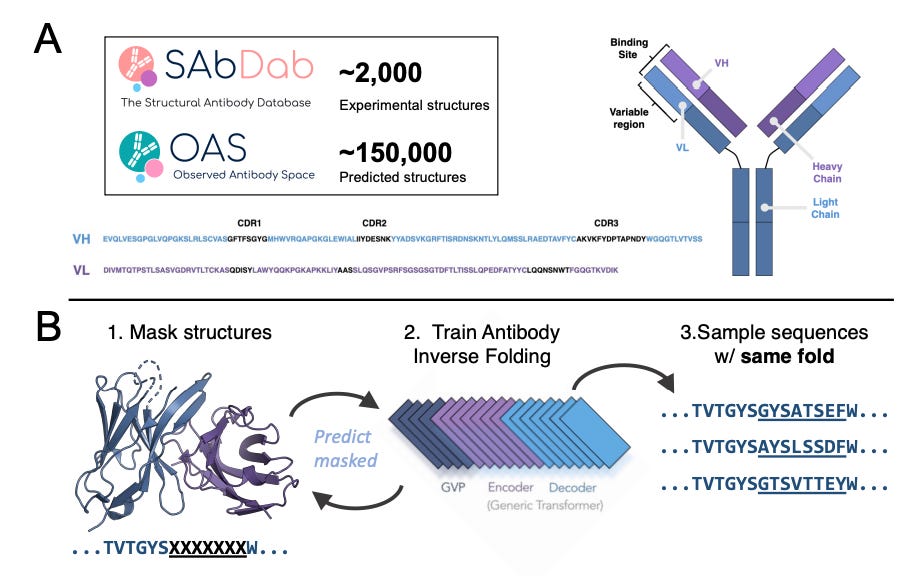
Given that some members of this team also developed AbMPNN, it’s a natural comparator for AntiFold. As demonstrated below, AntiFold outperforms both general and antibody-specific models in its ability to recover held-out, ground truth (read: experimental) amino acid sequences of known binders. AntiFold also appears to have improved perplexity when predicting amino acids. This means that the model suggests a narrower set of candidate mutations than AbMPNN.

Protein Design Using Diffusion Models
Diffusion models belong to the category of things I can’t believe work as well as they do. Crudely, diffusion models apply random (e.g., Gaussian) noise to a data distribution (e.g., an image) step-wise until it looks like pure static. The model then reverses this process, generating a final datapoint representative of the original distribution. Protein designers were creative applying diffusion models for a variety of tasks including motif scaffolding, protein-protein docking, and more.
A Framework for Conditional Diffusion Modeling with Applications in Motif Scaffolding for Protein Design
Kieran Didi spoke on his group’s work using diffusion models for generative protein design. Specifically, the team focused on motif scaffolding. Say you want to fix a specific, functional protein motif (e.g., an active site) and you’d like to build the rest of the protein around that motif—that’s motif scaffolding. To advance the field, the authors crafted a novel conditional generation technique, allowing them to steer the diffusion process towards a specific end state. Several concepts here are quite new to me, so I’ll do my best to faithfully represent what I learned.
As mentioned, diffusion models generate protein structures by learning to recover training examples that are iteratively noised with some forward process. The reverse process is where generation happens. Unconditioned diffusion models inefficiently sample the data distribution, as shown below on the left. Conversely, one can provide a signal to the model during the reverse process to coax it towards a specific final state. Kieran’s group developed amortized training, a conditioning technique that fixes the active motif and efficiently samples the data to arrive at a plausible scaffold.

The group’s conditioning method is based on Doob’s h-transform—a mathematical framework for steering a stochastic process towards a defined end state. The authors apply this approach during training, making conditional sampling during inference time more efficient. I won’t dig far into the math beyond the general form of the h-transform since (a) it’s discussed rigorously in the paper and (b) I’m still fully grokking it myself.

In the box above, X (Equation 1) is a time-denominated function that governs the reverse diffusion process—the generation of a protein structure from a fully noised state. Recall that time flows backwards from full noise (time (t) = 1) to final prediction at t=0.
X includes a drift term that guides that overall process and a diffusion term that adds or removes random noise.
With motif scaffolding, we’re trying to fix part of the final prediction. We want to adapt the model to generate structures that satisfy the condition where a specific active site is always in the final output, for example. Mathematically, that condition is expressed as X_o = B.
Doob’s h-transform is a way to condition a diffusion process on a desired, future state. To reflect the altered process, X becomes H (Equation 2), and includes a new term that guides the reverse diffusion process towards that final state B. The probability that H_o (the final output of the transformed process) arrives at the condition B is 1.
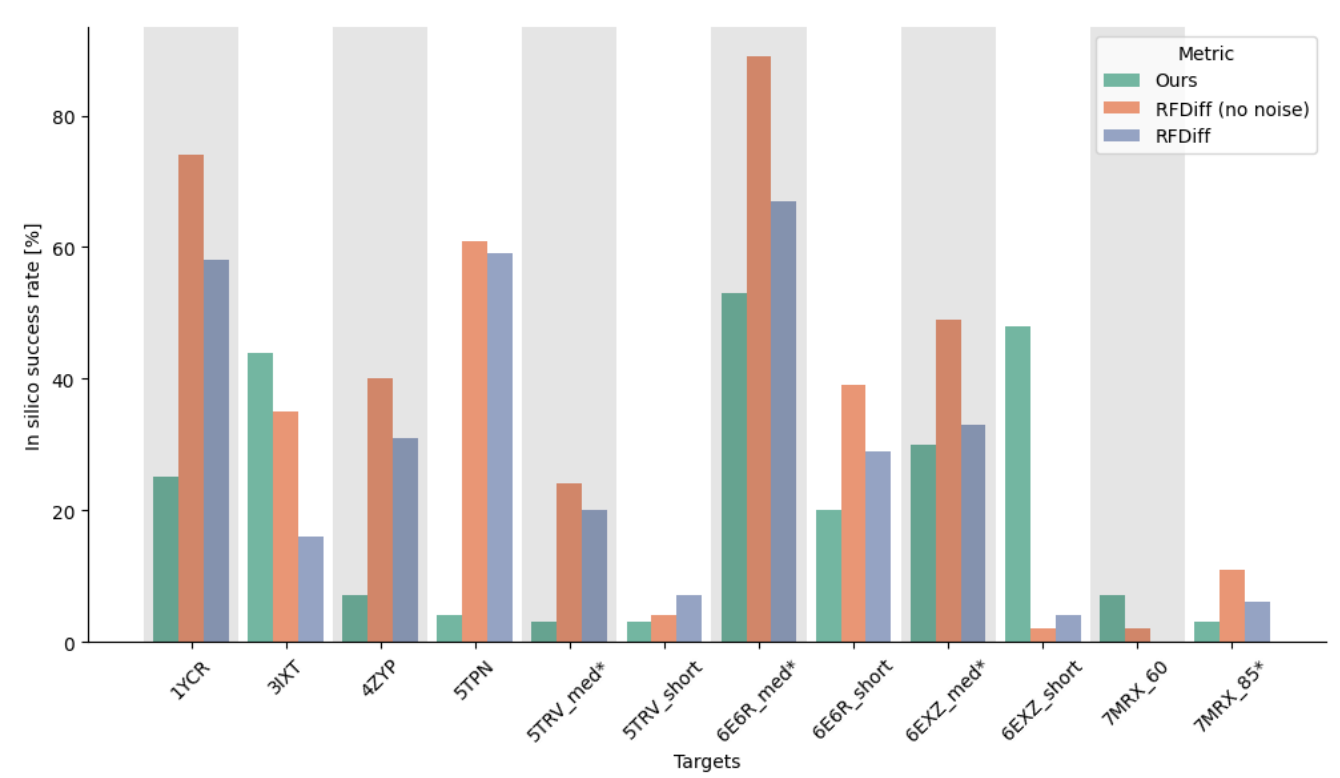
The authors compared this new approach against RFdiffusion on the basis of in silico design success rates across a dozen different proteins, as shown below. Though it underperformed RFdiffusion generally, the proposed amortized training technique is comparatively lightweight—requiring 10% of RFdiffusion’s parameter count and just 1.2% of the training time. Granted, in silico success isn’t an ideal performance benchmark—it’s mainly built on a <2 angstrom RMSD comparison to AlphaFold2. Regardless, this approach is extremely interesting. I’m eager to see how it performs in vitro once more developed.
Harnessing Geometry for Molecular Design
The inimitable Michael Bronstein presented on a host of papers, including the MaSIF family of geometric deep learning models. Hearing from Bronstein about the evolution of an idea over a five year period was a real treat. While this talk also featured fragment-based ligand design, I’ll save that discussion for my next post.
Published in late 2019, molecular surface interaction fingerprinting (MaSIF) is a framework for analyzing protein surfaces and predicting binding sites with ligands and other proteins. The main conceptual advance was focusing on protein surfaces and representing them as a patchwork of geometric meshes. As shown below, MaSIF pre-computes chemical properties of each patch, thereby co-embedding chemical and geometric features that contribute to binding another biomolecule.

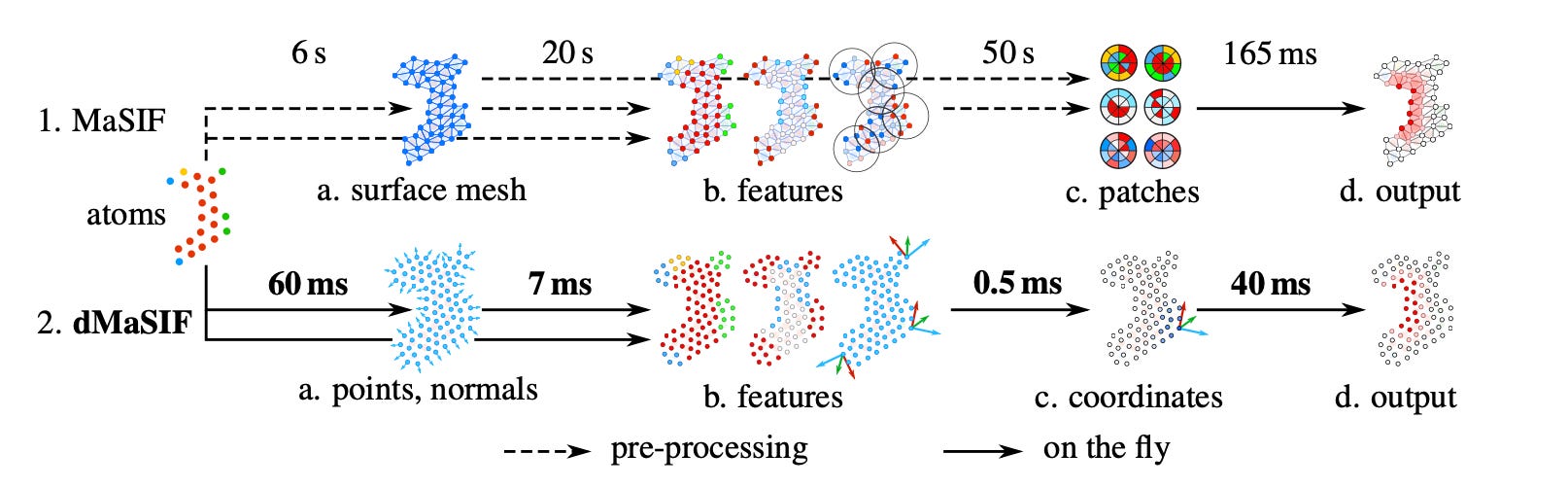
Though surfaces are efficient representations for learning intermolecular interactions, the authors cede that pre-computing chemical properties of patches is compute and memory intensive—requiring generation and storage of large batches of calculated data. MaSIF also has a limited ability to deal with conformational changes. This prompted the development of differentiable MaSIF (dMaSIF).
dMaSIF also focuses on surfaces, but uses a lightweight atomic point-cloud representation instead of discrete mesh patches. The key assumption is that a protein that’s sufficiently dense creates point clouds that aren’t that lossy compared to a mesh. Point clouds can be thought of as [x, y, z] coordinates that describe atoms’ positions in 3D space—a representation similar to that provided by x-ray crystallography. Unlike MaSIF, dMaSIF computes chemical properties on the fly directly on point clouds—this is much less compute and memory intensive, producing the speedup in the diagram below.
Diffusion MaSIF (DiffMaSIF) is a diffusion model for protein-protein docking that uses dMaSIF as its encoder, as shown in the diagram below. DiffMaSIF is purpose-built to work on non-native protein complexes. Normally, protein-protein models are trained on co-evolved protein sequences—complexes that evolved to fit together well. I wrote about this in a previous blog on the molecular dynamics of induced proximity drugs. Non-native protein interactions occur when designing proximity-based drugs like molecular glues—making DiffMaSIF highly therapeutically relevant.

My understanding is that DiffMaSIF embeds geometric and chemical properties of protein surfaces to predict likely binding sites (though the model can also be pre-conditioned on a binding site). Then, the decoder uses a form of score-based diffusion on the second protein—translating and rotating it on the first protein’s surface until a favorable binding orientation is found. Bronstein is Chief Scientist at VantAI, who likely uses this process in its pipeline for developing glue molecules. Dr. Correia (co-author) is also on the SAB, for transparency.
It’s standard to train on the Protein Data Bank (PDB) and test model performance on DB5, a subset of the Database of Interacting Proteins (DIPS) which removes proteins with >30% sequence homology. However, the authors use a more stringent test set called PINDER (see Appendix 3 in the paper). This helped mitigate the risk of data leakage and ensures the test set features a diverse array of protein complexes.

DiffMaSIF greatly improves the precision and recall of protein-protein binding site prediction over random surface sampling, as illustrated above. More importantly, it appears to outperform methods that use co-evolution of sequences across an array of protein-protein docking tasks. That said, DiffMaSIF still seems limited to rigid body docking, which isn’t representative of the diverse conformational landscape often occurring between non-native protein interactions. I imagine ways to handle this are on the horizon!
EDIT (1.21.24; 10:00 PM ET)—The authors did produce another manuscript about flexible protein docking. Check it out here.
De Novo Design of Protein Structure and Function with RFdiffusion
Developed by scientists at the Baker Lab, Brian Trippe and Jason Yim presented on one of the most well known generative protein models—RFdiffusion. I was fascinated to learn more about the backstory of RFdiffusion’s origin, especially in the context of RoseTTAFold. As shown below, standard RoseTTAFold uses a similar architecture to RFdiffusion.

The speakers elaborated on why base RoseTTAFold offers advantages as the neural network in a diffusion model. These included:
RoseTTAFold is rotation equivariant—the model understands if input data is rotated in 3D space and will adjust its output accordingly. This is useful since protein function is invariant to rotation, making the model more generalizable.
RoseTTAFold is a language transformer—it excels with sequential data (like strings of amino acids) and understands semantically meaningful shifts, insertions, and deletions of residues.
RoseTTAFold enforces some biochemical constraints—namely, it treats amino acid backbones as rigid, planar structures of atoms (N–Cα–C). This reduces the dimensionality (complexity) of the backbone representation and respects chirality (handedness) of the model, as shown below.
RoseTTAFold already has pre-trained structure prediction weights—making it easier to repurpose out-of-the-box for design tasks.

The speakers concluded by mentioning that RFdiffusion currently works with proteins only. Similar to many other generative models, RFdiffusion is not able to account for conformational dynamics. The next incarnation of the model, RFdiffusionAA, goes beyond residue-level protein representations to include discrete modeling of atoms, opening the door to many new applications including small molecule or nucleic acid binder design.
Protein Design Using Energy-Based Models
Energy-based models (EBMs) bridge energy and probability with how they represent data distributions. For example, proteins with favorable conformations exist in low energy states (local minima), increasing the probability that an optimization process lands there. The bridge between energy and probability is called the Boltzmann distribution.
DSMBind: SE(3) Denoising Score Matching for Unsupervised Binding Energy Prediction and Nanobody Design
Wengong Jin presented his group’s work developing DSMBind—an unsupervised energy-based model (EBM) for discovering protein binders. DSMBind is trained on the corpus of protein-protein (n=27,000) and protein-ligand structures (n=5,200) in the Protein Data Bank (PDB). DSMBind is an unsupervised model because training instances don’t have labeled affinity data. As a geometric EBM, DSMBind learns to minimize an energy function.
Assuming example crystal structures are at their lowest energy state to start, the authors apply random perturbations (noise) to protein backbones and side chains. This noise drives proteins to higher energy states—which DSMBind bind reverses using a process like gradient descent, as shown below in 2D.

Though based on an energy function, DSMBind is not a physics-based method. It’s not calculating a forcefield. DSMBind is rooted purely in statistical mechanics. The authors tested DSMBind on several tasks including protein-ligand binding prediction, protein-protein interface (PPI) mutation-effect prediction, and as a scoring function in a nanobody design pipeline.
They used 264 protein-ligand complexes from Merck and ~6,000 ΔΔG points from 348 protein-protein complexes in the SKEMPI database. Overall, they found DSMBind outperforms sequence-based (e.g., LLM) models and performs similarly to physics-based methods on these tasks, as shown below. Importantly, DSMBind made its predictions nearly four orders of magnitude faster than physics-based algorithms, perhaps enabling its use upstream for virtual screening.

For nanobody design, the team created two virtual screening pipelines utilizing DSMBind as a final scoring function. The first workflow (a, below) uses ESMFold to translate two nanobody sequence libraries into structures which are complexed with PD-L1 and scored using DSMBind. Using ESMFold was too slow, so the group created a lightweight (read: less accurate, but faster) version of DSMBind to narrow the sequence funnel. Finally, they tested two candidate variants experimentally with a negative control, as shown below.

Personally, it was extremely cool to see actual biophysical interactions manifest in DSMBind’s attention map. As shown below, this heat map lights up on ligand atoms that participate in a strong π–π interaction with protein binding pocket residues.

There were a handful of questions and comments, including some of my own. The diversity and representativeness of structures in the PDB are suboptimal for training generative models. How might this model perform on novel targets or epitopes less well captured in public databases? Most databases are enriched for binders, partially because display techniques eliminate negative training examples.
From an industry perspective, crystal structures take a fair bit of time to create. Would this hamper DSMBind’s utility as a virtual screening tool that is structurally enabled? Antibody design is a multiparametric optimization problem—binders need to be thermostable, pH-stable, non-immunogenic, etc. How could DSMBind be used alongside other property-specific algorithms?
Thanks for reading and be sure to subscribe below if you want to read Part 2—Generative Chemistry. Did I get anything wrong? Do you have takes either way on any of these methods? Feel free to comment!
—Simon Barnett, Head of Research