

The Isoform Frontier
Our ability to cost-effectively analyze mRNA isoforms at scale will bring profound advancements to basic and translational research. The added resolution may unexpectedly boost proteomics as well.

Gene expression profiling is undergoing another technological renaissance. In the late 1990’s, the Affymetrix Gene Chip enabled genome-wide expression profiling. One decade later, next-generation sequencing (NGS) supercharged RNA expression analysis by offering substantially better throughput and economics with RNA-Seq.
Improvements in scalability were soon followed by advancements in resolution. Nowadays, researchers can profile gene expression at the single cell level and increasingly with sub-cellular spatial resolution in intact tissues. Recent papers have even proposed methods to infer temporal gene expression dynamics using Raman spectroscopy and generative adversarial networks (GANs).
Though standard gene expression profiling will remain a foundational pillar of molecular biology, we know that genes aren’t expressed—full-length RNA transcripts (isoforms) are the actual molecules being translated into proteins.
The field’s lens into the transcriptome is poised to sharpen once again. I believe the ability to accurately sequence full-length isoforms cost-effectively and at scale is a transformational improvement. The impacts could be numerous and far reaching. These include functional genome annotation, biomarker discovery, cancer research, drug target identification, and more. I’ll save application-specific deep dives for another time and instead focus on three things:
How does isoform sequencing shed light on elusive, high-value biological mechanisms?
What technological breakthroughs are enabling isoform sequencing?
How can isoform sequencing create exciting, non-obvious improvements in proteomics analysis?
Single genes can create many different proteins. Sometimes, proteins encoded by the same gene are just as different as proteins encoded by different genes. To understand how this happens, let’s first zoom in on transcription—the process of copying a segment of DNA (a gene) into RNA.
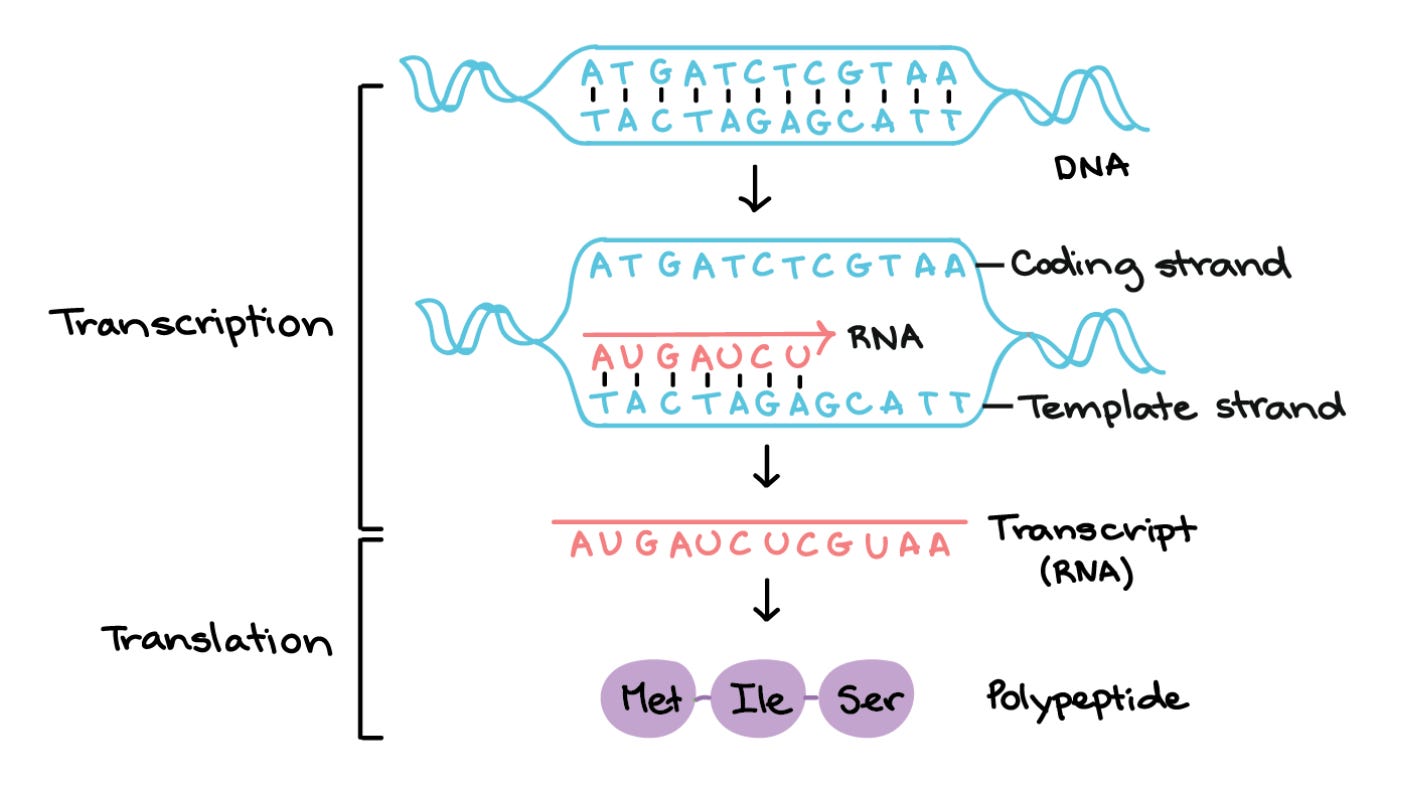
Recall that genes are composed of coding and non-coding motifs called exons and introns, respectively. Young RNAs (pre-mRNAs) contain many exons and introns. Before they’re translated into protein, our cells remove a pre-mRNA’s introns and stitch exons together. Alternative splicing refers to the different ways exons can be included or excluded in a mature mRNA molecule. Intuitively, mRNAs with different combinations of exons (isoforms) will translate into different proteins, as shown below.
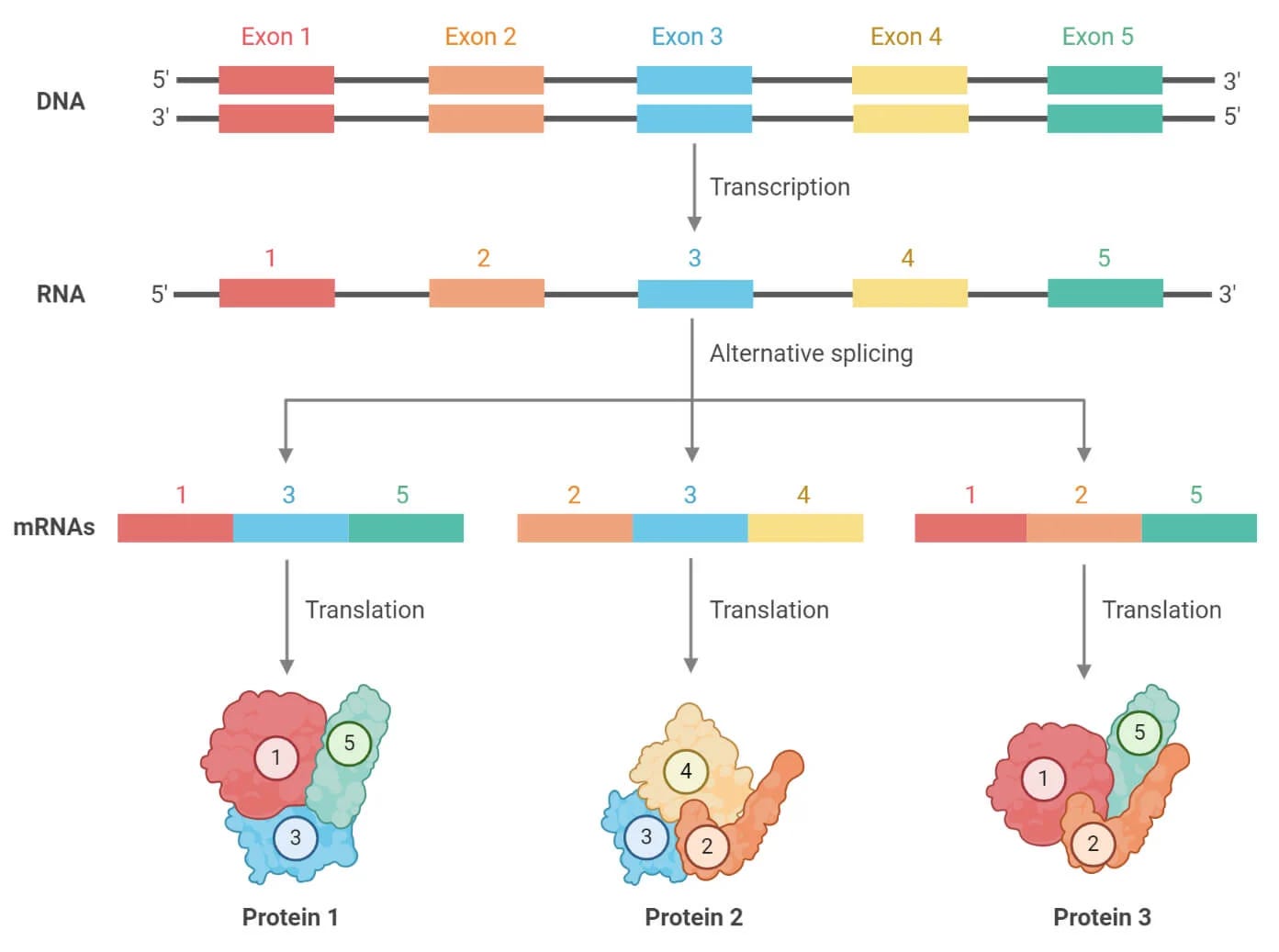
Alternative splicing is just one of many transcriptional mechanisms explaining how many proteins can come from a single gene. Alternative splicing is ubiquitous, affecting 95% of human genes and occurring in nearly all organs. Splicing (and mis-splicing) is especially common in neurons and in solid tumors and has been linked to myriad diseases. These facts make splicing a crucial phenomenon to understand. Therefore, they also make isoform sequencing a crucial method to deploy.
NGS is the bedrock of RNA sequencing. It’s the readout platform of choice for bulk, single cell, and even spatial analysis of gene expression. Unfortunately, RNA-Seq has limited utility for studying RNA isoforms for the simple fact that short reads are 50-600 base pairs (bps) long while full-length human isoforms measure ~1.6 kbp.
As sequence reads accumulate on a particular transcript, we can measure the level of expression of its parent gene. However, getting isoform-specific expression data is tricky unless a read happens to span a canonical splice junction. Even so, some isoforms are distinguished by several unique splice junctions separated by hundreds of base pairs.
This challenge has spawned a wave of innovative chemistry and algorithmic hacks to improve isoform resolution with NGS like Smart-Seq3 and Kallisto, respectively. These protocols are powerful, but still struggle to precisely resolve all human transcript isoforms. In my view, this ambition requires sequencing methods capable of generating long (>5kb), high accuracy (>99% or Q20+) reads at high throughput (>2x10^8 reads) and low cost (<$10/Gbp). Until recently, no technologies satisfied all these criteria.
Over the past 18 months, long-read isoform sequencing workflows from Pacific Biosciences (PACB, PacBio) and Oxford Nanopore Technologies (ONT.L, Nanopore) have breached these criteria. According to recent filings, these technologies are the fastest growing in the sequencing marketplace, meaning the capacity to resolve isoforms is growing rapidly in turn. My current opinion is that RNA-seq will remain the workhorse assay for transcriptomics, but that long-read isoform sequencing will proliferate rapidly and confer novel insights to its adopters.
As mentioned, the recipe for isoform sequencing calls requires (a) high-accuracy, (b) high-throughput, (c) long read lengths, and (d) low costs. For years, PacBio and Nanopore platforms checked several—but critically not all—of these boxes. A handful of dam-bursting technological improvements have struck since.
Nanopore solved throughput and cost with the 2019 launch of its PromethION flow cell which can produce hundreds of Gbps per run at a <$10/Gbp price-point. Users can run from one to 48 PromethION flow cells in parallel on the lightweight P2 or beefy P48, respectively. Nanopore sequencers aren’t short or long-read—they’ll sequence whatever length DNA or RNA one feeds into the system, including 1.6kbp isoforms.

Accuracy was the rate-limiting ingredient for isoform sequencing using Nanopore systems. For years, Nanopore’s modal accuracy peak lived in the range of 90-99% (Q10-Q20), as shown below. This made it challenging to confidently demultiplex barcodes attached to DNA molecules when batching multiple samples together. Recently, Nanopore launched its V14 chemistry built atop a better signal from a new pore design (R10.4.1) and a refreshed deep learning model for basecalling, raising its raw accuracy to >99%—thus mitigating its Achilles heel and positioning its technology as an excellent solution for both bulk and single-cell isoform sequencing.

PacBio solved accuracy in 2019 with the release a circular consensus sequencing (CCS, a/k/a HiFi). This method combines sub-reads from the same original molecule, generating consensus read accuracies above 99.9% (Q30). HiFi reads generally fall within the range of 15-20 kbp, certainly long enough to sequence isoforms.

Cost and throughput were PacBio’s rate-limiting ingredients. The company’s previous platform (Sequel IIe) could generate just 2.5 million reads per run at a cost of $550/million reads. This year, PacBio launched two products that, when combined, increase throughput to 320 million RNA (technically cDNA) reads per run at a cost of $13/million reads. These products include a new flagship platform (Revio) and a purpose-built isoform sequencing chemistry (MAS-Seq) originally developed at the Broad Institute.

Altogether, long read platforms now check all the boxes and are diffusing into the marketplace quickly, setting the foundation for ubiquitous isoform discovery. It will take time for novel discoveries to trickle into the corpus of scientific knowledge. Until then, I want to highlight a specific and maybe non-intuitive application—combining long-read isoform sequencing and mass spectrometry to supercharge protein identification.
Proteomics represents the next mysterious frontier of the ‘omics stack. Our arsenal for unraveling the proteome is underpinned by mass spectrometry (MS) and affinity reagents (e.g., antibodies, aptamers). Despite the prospects of truly novel protein sensors—which I think are necessary and inevitable—I believe MS and affinity-based methods will remain the primary assays for years to come. As such, the combination of MS and rapidly expanding isoform sequencing capacity is apt to occur.
MS involves digesting proteins into small peptide fragments and shooting them through the instrument to produce mass spectra. These spectra feature a series of spikes that correspond to the mass-to-charge (m/z) ratio of peptides in the sample. Researches compare these spectra to those contained in reference databases to identify which peptides are in their sample. Inferring full-length proteins (proteoforms) can be challenging since some peptides can map to multiple proteins. Sometimes, groups will assign peptides to protein groups or families as a compromise.

Theoretical peptide databases may not adequately represent proteomic variation across tissue types or disease states, complicating analysis. Meanwhile, sample-specific databases of full-length RNA isoforms help researchers know what proteins could exist in a sample. Unsurprisingly, many have applied RNA-seq to build sample-specific databases. As mentioned, these techniques cannot adequately resolve the molecules in a sample actually being translated—mRNA isoforms, not genes.
Last year, the Sheynkman lab published a paper and open-source Nextflow pipeline for combining long-read isoform sequencing and mass spectrometry. Her team’s work highlights the advantages of such an approach for cataloguing the cellular proteome. I’ve pulled out a few highlights below, though I recommend reading the whole work:
By incorporating isoform sequencing, Sheykman’s group surfaced ~45,000 candidate protein isoforms from a well studied human cell line (Jurkat). Nearly 23,000 (50%) were novel.
75% of human genes generated a novel, not-in-catalog protein isoform. For a third of human genes, the most abundantly expressed protein isoform did not match the most abundant reference isoform.
The isoform-resolved method recapitulated 99% of peptide-level and gene-level identifications when compared to a popular MS database (GENCODE). However, only 41% of protein group-level identifications overlapped—illustrating how proteoform classifications can vary widely based on the isoform compositions of the MS reference database.
Combining RNA isoform sequencing with MS is a powerful approach to detect protein isoforms resulting from genetic variation and post/co-transcriptional mechanisms such as alternative splicing. Because long read sequencing and MS already have broad adoption, workflow support, and cost-effectiveness, the combination of these approaches is poised to provide near-term impact on basic and translational proteoform discovery.
Today, a common approach linking RNA and full-length proteins is single-cell CITE-Seq which combines RNA-seq and fluorescent antibody readout. This method is unable to detect RNA isoforms and is limited to surface proteins. Conversely, proteogenomics methods combining MS and isoform sequencing provide a lens into intracellular proteins and RNA isoforms alike. To be fair, single-cell proteomics with MS is challenging. Though I’m excited by recent improvements in MS sensitivity, it remains a targeted method.
Proteogenomics using MS and isoform sequencing has its limitations. Using this approach, researchers would be able to tell which exons are expressed and whether they co-exist on specific mRNA isoforms, but exact quantification of the resulting proteoforms would remain elusive. This method would not provide an advantage with the detection of post-translational modifications (PTMs) either. For these reasons, I’m still convinced that we’ll need a new sensor that can tackle the massive molecular diversity, dynamic range, and information content of the proteome.
What’s a technology article without a bit of speculation? As mentioned earlier, the rapid adoption of third-generation sequencing platforms (e.g., long reads) will catalyze many areas of discovery. I predict this class of sensor will generate >$1 billion in sales within the next few years as it saturates new labs and takes on larger projects.
We will see a major boost in our ability to see entire genomes and assign functions to genes. Generating data on multiple facets of biology—DNA, methylation, chromatin accessability, transcriptomics—these will become standard from every sequencing run. Novel biomarkers derived from previously unseen processes like alternative splicing or obscure variant classes (e.g., tandem repeats) will spur therapeutic inquiry. Domains like immunology, cancer, rare disease, and neurology will advance in bounds.
If you have conviction over a particular biosensor x application area combo, feel free to DM me on Twitter or leave a comment. Dimension is steadfastly interested in any and all tools capable of systematically eliminating the Streetlight effect within molecular biology.
Simon - Thanks for writing this. Very helpful. My friend at U Penn, Ian Blair, is using targeted mass spec to elucidate the proteoforms in different settings. For example, the genetic disease, Friedreichs Ataxia (FRDA), where he discovered a novel splice variant of the deficient frataxin protein. Even "simple" genetic diseases appear more complex than anticipated, and such insight is vital for developing effective therapies. Ian has also done seminal work on clarifying the proteoforms of the cryptic molecule HMGB1. Unfortunately, the HMGB1 literature is contaminated with incompetent and fraudulent mass spec which has caused immense confusion. Mass spec proteomics as done by Ian has a huge amount to offer right now. Kevin