

An Opinionated AlphaFold3 Field Guide
I break down every part of AlphaFold3 in simple-to-understand terms and give my observations and opinions along the way.

Intro & Main Takeaways
AlphaFold3 (AF3) represents another stellar leap in biological capability afforded by machine learning (ML).
The headlines are that AF3 is more accurate, generates structures with greater resolution, and is compatible with molecules beyond proteins. In this essay, I break down each aspect of the model in lay terms and give my current observations and opinions on AF3. Let’s start with the main takeaways:
Unlike its predecessor, AF3 is a generative model. This means it can give different outputs for the same input and that it can hallucinate, a phenomenon evident with intrinsically disordered regions (IDRs) of proteins.
DeepMind & Isomorphic squeezed all the juice out of the Protein Data Bank (PDB), highlighting the paucity of training data in the life sciences. Incorporating protein dynamics or multi-parameter optimization are likely next steps.
The era of vast improvements in structure generation using open-access data repositories seems to be coming to an end. Proprietary data generation will be key to unlocking the next 10x improvement in ML for molecules.
The lack of code and constrained usage server make AF3’s claims hard to replicate or verify. Until an open version is built, it’ll be hard to tell how AF3 generalizes.
By removing equivariance from the model, AF3 has agitated the debate around the best path forward for capturing physical invariances. I explain this more simply in the essay.
We’re still a long way from replacing experimental methods with ML in the life sciences. That said, progress has been breakneck this decade and one day when I write one of these articles—ML will have replaced some experimental methods.
AlphaFold3

AF3 is trained to generate 3D structures of molecules input by a user. Having a viable 3D structure aids hypothesis generation and is a prerequisite for rational drug design.
In summary, users input raw data files containing the molecules they want to combine into a structure. The model processes these data into a format that the main neural network can operate on efficiently. AF3 leverages certain databases to extract structural and co-evolutionary information that can help inform the model. The model then generates a structural hypothesis that’s subsequently handed off to a generative module, called a diffusion model, which generates the final, 3D atomic structure. AF3 also outputs several quality metrics that help users understand the likelihood that a structure is correct.
Let’s dive into the first part of the model—the raw input layer.
Raw Input
Compared to its previous generation, AF3 handles many more types of molecules This necessitated some major changes to the model.
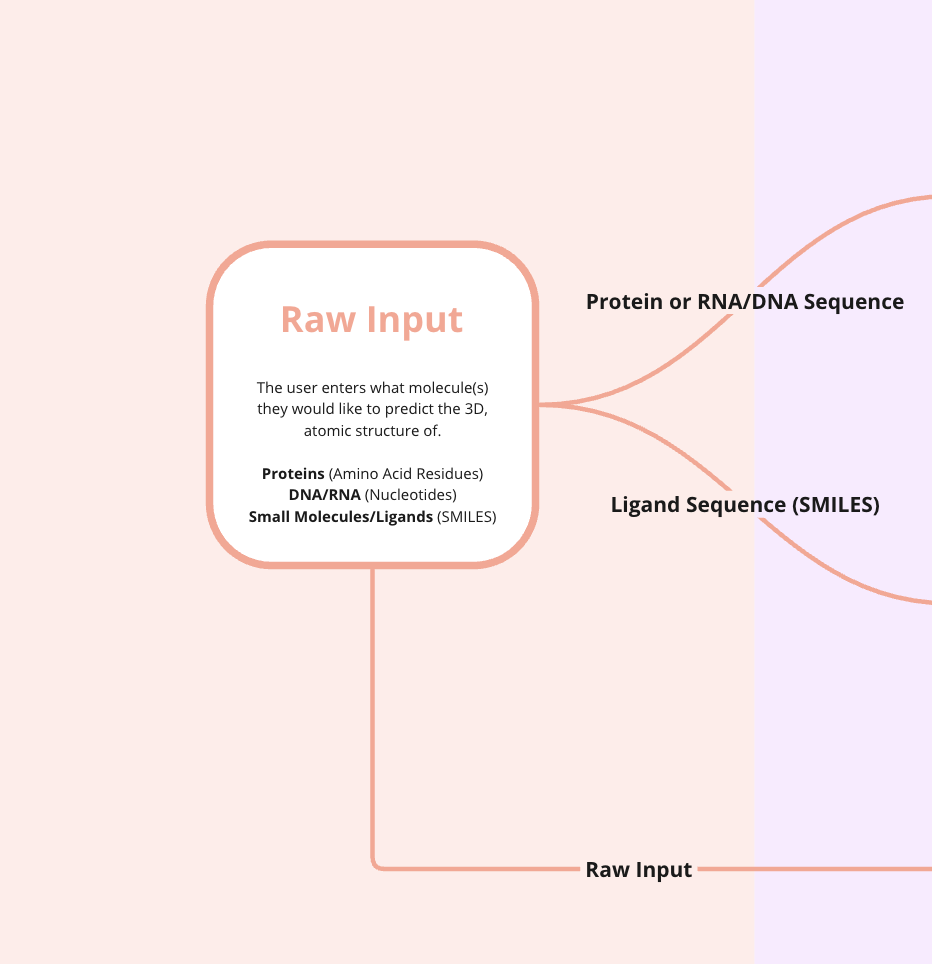
The first stage of AF3 involves users inputting raw data. These data describe the molecules AF3 will attempt to generate structures for. Unlike AlphaFold2 (AF2), AF3 is capable of handling a more diverse set of molecule types, including proteins, nucleic acids (e.g., DNA), and small molecules (i.e., ligands).
Users can input one-dimensional (1D) sequences of molecules. This is straightforward for proteins and nucleic acids since they can be represented as strings of subunits. For ligands, users can submit SMILES strings—a naming convention that encodes 2D molecular structure in a 1D text string.
Data Preprocessing Pipeline
AF3 cannot operate directly on raw data. Input molecules must first be transformed into a format that a neural network can manipulate. AF3’s data preprocessing pipeline is the first step in the conversion process.

AF3’s data preprocessing pipeline is more complicated because the model handles so many molecule types. Harmoniously combining them in a single, network-interpretable format is a tall order.
Conformer Generator
This block receives a ligand in the form of a 1D SMILES string and converts it into a 3D structure called a conformer. DeepMind didn’t reinvent the wheel. The Conformer Generator uses ETKDG (v3), a tool contained within a popular, open-source cheminformatics software suite called RDKit. ETKDG is optimized for small molecules. It’s possible this module becomes less effective with larger molecules (e.g., natural products).

Genetic Search
This block receives a protein or nucleic acid sequence—the query sequence. Using the query sequence, the Genetic Search module surfaces similar, highly-overlapping sequences found in nature. Each of these similar sequences is stacked below the query sequence in a chunk of information called a multiple sequence alignment (MSA).
The MSA is an MxN matrix—a grid of values that has M rows and N columns where N equals the length of the query sequence and M equals the number of unique sequences gathered from the database.
")
Amino acids conserved across organisms are generally structurally or functionally important. Co-evolutionary sequence information enables AF3 to infer which amino acids contact each other, assisting the structure generation process as shown below.

Template Search
This block receives the MSA and outputs a set of candidate protein or nucleic acid structures that downstream parts of AF3 can use to guide the structure generation process.
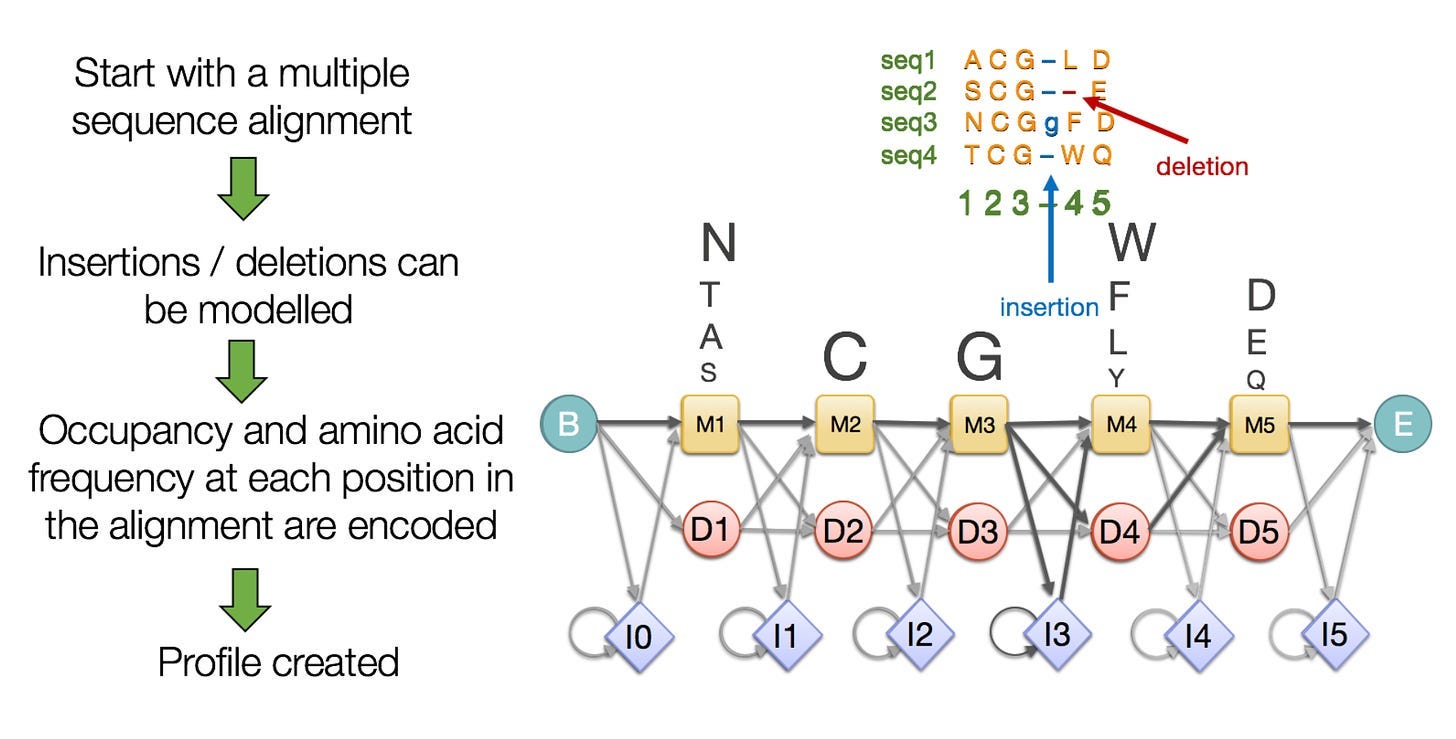
The template Search first trims the MSA to include only the 300 most relevant sequences. The module then transforms the MSA into another type of data object called a Hidden Markov Model (HMM).
HMMs are adept at flexibly representing sequences with divergent regions—where parts of a protein are flipped, deleted, or inserted. Each amino acid position is given a score according to the frequency it occurs in the MSA. HMMs also incorporate probabilities, such as the likelihood that a specific amino acid follows another specific amino acid. Read more about HMMs for proteins here.
Using the HMM, the Template Search extracts up to four candidate structures from a database. Like the Genetic Search, the Template Search provides structural cues that can assist AF3 in generating the final structure.
At the end of the data preprocessing pipeline, we’re left with a 3D ligand structure, an MSA, and a set of structural templates. We also still have the original, raw input. Unfortunately, none of these data objects are fully compatible with the neural network. This is because the data are currently in the form of one-hot encodings rather than continuous embeddings. Transforming into the latter format requires the embedding layer.
Embedding Layer
This segment of AF3 is responsible for converting raw structural and biological data into a mathematical representation that the main neural network can act on directly. This layer also enriches embeddings with structural and co-evolutionary insight.

I’ve included the Input Embedder, Template Module, and MSA Module in what I’m calling the embedding layer. This is a subjective. Because the Template and MSA modules are additionally tasked with extracting structural and co-evolutionary information and because these blocks are involved in a recycling loop, it’s tempting to include them in a subsequent section.
I’ve chosen to lump them into the embedding layer because I wanted to write about the structure generation layers separately. Let’s start with some relevant definitions:
Tokenization involves breaking apart big pieces of data into bite-sized chunks called tokens. AF3 tokenizes molecules with varying degrees of granularity. Proteins and nucleic acids get tokenized as amino acid residues and nucleotides, respectively. Ligands get tokenized atom by atom. Tokens still exist in data space—they’re interpretable by humans.
Embedding means converting discrete tokens into continuous, mathematical objects like vectors and matrices. Continuous embeddings facilitate matrix multiplication, a common operation within neural networks. The following image illustrates tokenization and embedding for natural language, but the process is similar for decomposing strings of molecules.
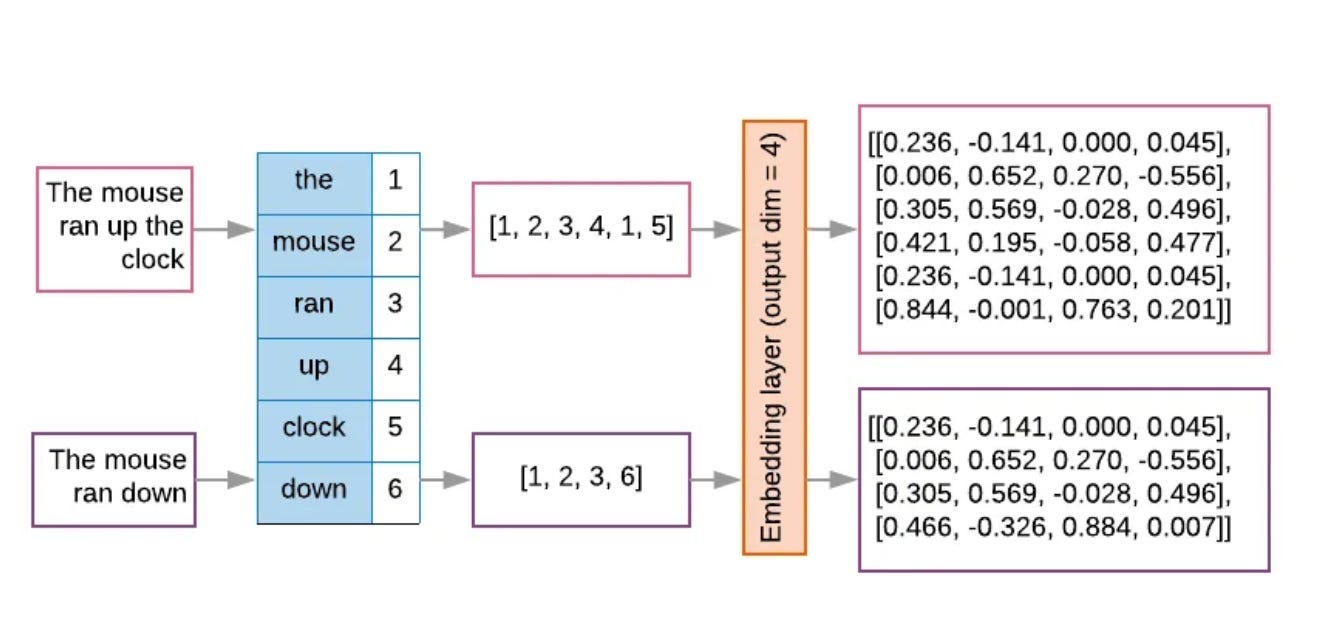
The original sentences and initial tokenization T both exist in data space. The orange block converts these structures into embeddings that an ML model can use directly. (Source) Attention is an ML mechanism whereby the model pays more focus to certain parts of its input data. This is accomplished through attention weights—amplifying the importance of certain data.
Now let’s get back to the model.
Input Embedder
This block is the embedding workhorse. It receives the original, raw input data, the 3D ligand structure from the Conformer Generation block, and the original MSA from the Genetic Search block. The Input Embedder produces three embeddings: an input representation (IR), a single representation (SR), and a pairwise representation (PR).
Input Representation (IR)
The IR contains high-level information about the raw input. First, the Input Embedder converts every atom into a token and embeds those into a vector. Per-atom features also include things like atom type, position, charge, etc. The module adds (concatenates) and embeds tokens for every amino acid or nucleotide. Therefore, the IR contains comprehensive, basic information about all atoms and residues.
Pair Representation (PR)
While proteins and DNA/RNA can be represented as linear strings of characters, they’re actually dynamic, 3D objects. To capture spatial relationships, the Input Embedder lays all the atoms flat on a table and allows them to talk to one another. Using a process called sequence-local atom attention, the model focuses on local neighborhoods of atoms and learns general rules about how these atoms might later interact with another to form a stable 3D structure—producing the PR.

Laid out in an all-by-all grid, sequence-local atom attention allows the model to give closer attention to the yellow-highlighted regions, local pockets of atoms. (Source)
Single Representation (SR)
The SR is an extension of the original IR. Here, the Input Embedder groups atoms depending on the nucleotide or amino acid they belong to, adding more context and meta-data. Though technically independent, atoms in the same functional unit (e.g., amino acid) behave differently than free-floating atoms.

Template Module
This block receives both the PR from the Input Embedder as well as the 3D template structures directly from the data preprocessing pipeline. The Template Module’s job is to refine the PR by incorporating structural information about closely related proteins found in nature. This allows the network to focus its attention on specific regions in the template based on the model’s current belief about the structure.
MSA Module
This block receives the template-augmented PR output from the preceding Template Module as well as the IR and the raw MSA from the Genetic Search module contained in the data preprocessing pipeline. This module’s job is to extract the co-evolutionary signal from the MSA into the PR, helping the model focus its attention on recurring sequence motifs.
At the end of the embedding layer, our raw data have been transformed into token embeddings that capture the identity, properties, and spatial interdependencies of atoms and other salient functional units (e.g., amino acids) of molecules. These embeddings exist in a format that the main processing trunk of AF3 can operate on directly. We won’t see information in data space—human-interpretable space—again until the final model output.
Pairformer
The Pairformer stack is the central trunk of AF3. It generates a structural hypothesis that downstream parts of the model can leverage. At a high level, the Pairformer takes in unrefined embeddings, processes them, then recycles those results a number of times to improve accuracy and performance.

This block receives the PR after it’s been enriched with template and co-evolutionary sequence information. The Pairformer also incorporates the SR and the initial input embedding. Following 48 recycling iterations, this module refines its belief about the structure—which it outputs in the form of a refined PR and SR.
Importantly, the Pairformer operates in latent space—the mathematical Twilight Zone. When I say “3D” in this section, I’m referring to the x,y,z-directions in a matrix of numbers—a lower-dimensional embedding space.

Part of Pairformer’s job is to determine structure by determining whether data correlations are due to physical contact or are simply co-evolutionary. Using a familiar diagram, notice that an observed data coupling between residues i and k can be a result of close contact between i-j and j-k—not necessarily that i-k are physically connected. Discerning real contact from observed correlation requires some sort of inference.

Pairformer treats structure generation as a graph inference problem in “3D” space. The model tries to understand how tokens influence each other in the latent space using rules of geometry. Specifically, it invokes the triangle inequality—that the sum of any two sides of a triangle is greater than or equal to the third side. Imposing this principle on the model is a human heuristic hard-coded into the network—an inductive bias.
Three tokens makes a triangle in “3D” space. Using i, j, and k from earlier, Pairformer performs an iterative series of triangular updates between each token—a point (node) in the triangle. Updates are mathematical changes to the PR that reflect the nature of the relationship between i, j, and k. Pairformer then leverages an attention mechanism to determine which sets of tokens are most relevant for building out its final structural hypothesis. Via a 48-times-refined PR and SR, the Pairformer stack hands off the data downstream to the diffusion module.
Diffusion Module
The diffusion module is responsible for transforming refined Pairformer embeddings into true-3D coordinates of atoms.

Diffusion is an established technique in the molecular sciences. I discussed diffusion models for generative protein design in a previous essay.
Diffusion models are a class of generative ML model. During training, diffusion models apply usually-random noise to a distribution of data until it resembles pure static, for example. The model then learns to reverse this process, generating final data points representative of the original data distribution (see below). Diffusion models excel at generating samples from very complex, underlying data as found in the life sciences. AF3’s diffusion module generates what it believes is the correct true-3D structure of a user’s input molecules.
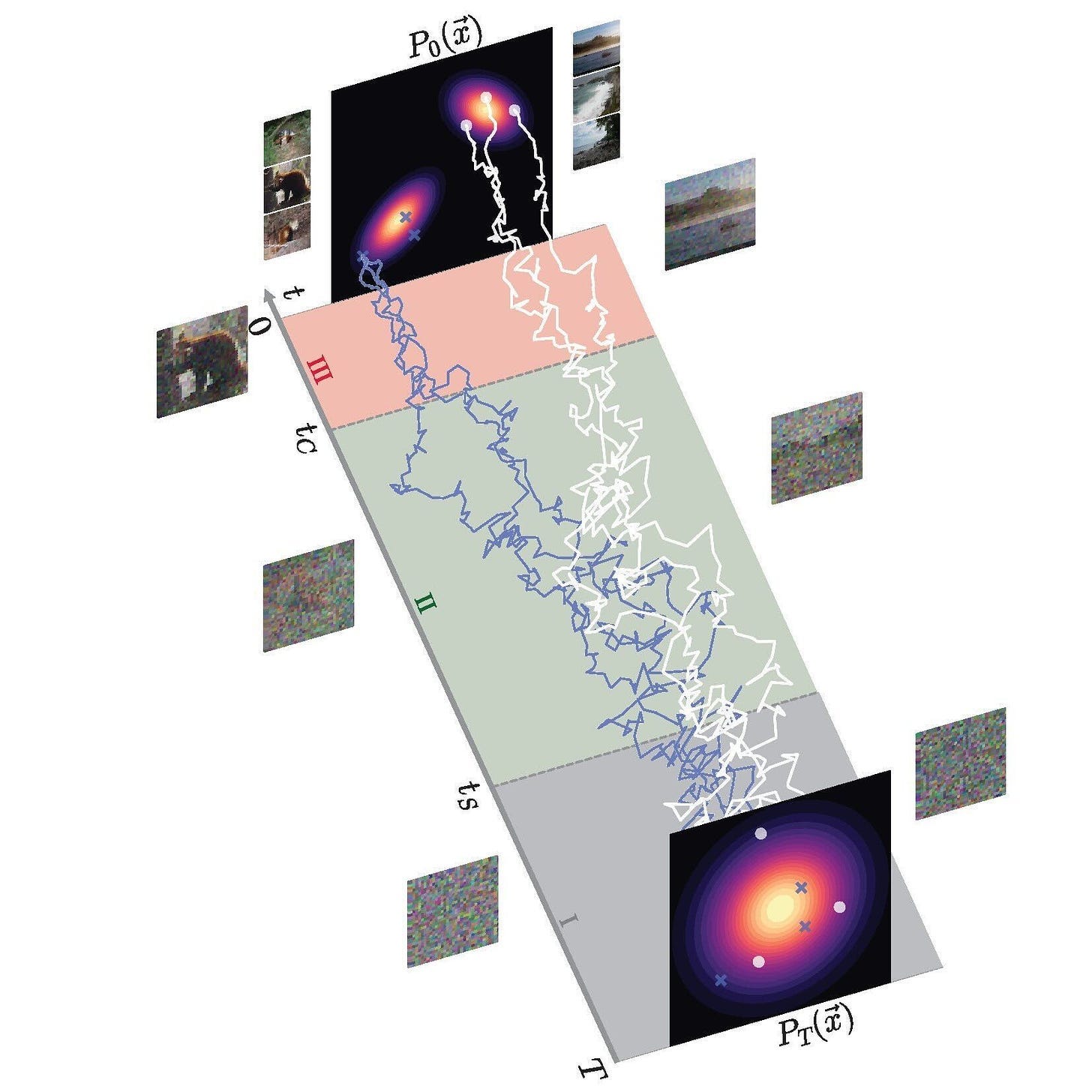
Diffusion models don’t always infer efficient paths from noised to final data. The trajectories are jagged. Conditional diffusion is a method to make diffusion more efficient. AF3 uses this mechanism by giving the Pairformer hypothesis to the diffusion module—essentially guiding the diffusion process. Using this structural hint, AF3 can efficiently generate accurate, 3D structures. That said, the diffusion module doesn’t always produce the same output for a given input. Like other generative models, diffusion models can hallucinate—more on this later.
Confidence Head
The final stop on the AF3 journey, the Confidence Head outputs a series of scores meant to help users understand the model’s confidence about the structure’s accuracy. Confidence metrics help biologists have faith in a model’s output as they’re often correlated with true accuracy.

The Confidence Head receives the three main model embeddings (IR, SR, and PR) as well as the final 3D structure generated by the diffusion module. Leveraging these, the Confidence Head generates a series of confidence metrics, which I cover below.
Predicted Local Distance Difference Test (pLDDT)
This metric gives a score (0 to 100) that reflects how confident the model is about local parts of its structure. Essentially, it colors a molecule with a heat map so users understand areas of local confidence. This is highly relevant for certain protein motifs that are functional like active sites.
Predicted Aligned Error (PAE)
The PAE estimates the error for each residue (e.g., amino acid) in the generated protein structure if it were aligned with the corresponding residue in the ground truth structure.
Predicted Distance Error (PDE)
In addition to the error flowing from alignment issues, the model also predicts mistakes in its ability to generate atoms that are correctly distance from one another in 3D space.
Experimental Resolution Test
This test measures whether particular atoms in the generated structure are resolved in the known, ground truth structure or not. Recall that ground truth structures are generally determined experimentally via x-ray crystallography.
Observations & Opinions
Generative; Not Predictive
“Unlike its predecessor, AF3 is a generative model. This means it can give different outputs for the same input and that it can hallucinate, a phenomenon evident with intrinsically disordered regions (IDRs) of proteins.”
I’ve intentionally used “generative” instead of “predictive” throughout this piece. Unlike AF2, AF3’s diffusion module make it generative and occasionally hallucinatory—it can generative deceivingly good, yet inaccurate structures.
Intrinsically-disordered regions (IDRs) of proteins are one are where AF3 is prone to hallucinate. AF3’s developers used a cross-distillation strategy to remedy this issue. They trained AF3 using IDR protein structure predictions from AF2. As a predictive model, AF2 had a strange, useful tendency to generate ribbon-like structures within IDRs. The cross-distillation strategy encourages AF3 to adopt this behavior instead of forcing a low quality structure into an IDR.
A Logical Step; But What’s Next
“DeepMind & Isomorphic squeezed all the juice out of the Protein Data Bank, highlighting the paucity of training data in the life sciences. Incorporating protein dynamics or multi-parameter optimization are likely next steps”
We knew for certain when DeepMind & Isomorphic published the AF-latest article that AF3 would include molecules beyond proteins. This is logical since the PDB contains proteins bound to a plethora of things—small molecules, nucleotides, random biomolecular shrapnel, etc.
AF3 is likely >6 months behind DeepMind & Isomorphic’s internal state-of-the-art (SOTA). They’re probably parallel-tracking many avenues of improvement. Two in my mind are incorporating protein dynamics and/or conformational flexibility as well as adding multi-parameter constraints to the diffusion model.
Protein complex are always vibrating, twisting,, and contorting through time. Many industry-relevant tasks hinge upon this dynamical understanding of a target protein. Since AF2, researchers have hacked the model to sample ground-state protein conformations (e.g., with AF-Cluster). Recently, models like AlphaFlow have produced short-lived, equilibrium thermodynamic fluctuations around protein ground states. In a soon-forthcoming article, I’ll advocate for neural network potentials here. Now switching gears.
Drug development is inherently multi-parameter—one cares about efficacy and safety and developability. Constraining diffusion trajectories to satisfy all of these objectives could be another direction DeepMind & Isomorphic take.
Data is Diamonds
“The era of vast improvements in structure generation using open-access data repositories seems to be coming to an end. Proprietary data generation will be key to unlocking the next 10x improvement in ML for molecules.”
Leo Wossnig and Winston Haynes covered brilliantly the importance of data quality and quantity for training performative ML models. Models are only as good as the data they’re trained on. The AF series has been successful because the PDB is an immense dataset. However, we seem to be reaching the era of diminishing returns—and there’s not another dataset like the PDB laying around.
I’m not of the belief that a new ML architecture or training method will confer another order of magnitude improvement for structure prediction and/or generation. My strong sense is that proprietary data generation, even if used for fine-tuning, is the path forward for companies like DeepMind & Isomorphic. This has implications on how the field will evolve if SOTA models increasingly hail from those capable of paying for immense data generation.
A Posture Shift
“The lack of code and constrained usage server make AF3’s claims hard to replicate or verify. Until an open version is built, it’ll be hard to tell how AF3 generalizes.”
The launch of AF3 didn’t look like the launch of AF2. There will be a 6-month waiting period before the community gets access to the code behind AF3. Inferencing the model requires a web portal. Use is constrained to non-commercial applications. There was no ablation study in the paper that highlights the importance of each component of the model. I acknowledge that there was a kerfuffle on Twitter (X) about all this.
Honestly, it makes sense to me. Isomorphic put out well-commented pseudocode and opened the model to some extent for others to use, which should accelerate open-source development. This is much more than the average, closed source computational biology company. Isomorphic has been clear and consistent in its communication that they’re iterating towards becoming a vertical biotech with a pipeline. I don’t want to belabor the point, but I felt the need to at least acknowledge it.
Equivariance is Dead; Long Live Equivariance!
“By removing equivariance from the model, AF3 has agitated the debate around the best path forward for capturing physical invariances. I explain this more simply in the essay.”
This section could be a blog post on its own—and likely better written by someone with a deeper knowledge of equivariance, but I will do my best.
One major argument that broke out post-AF3 was about the model jettisoning the concept of equivariance in the main structure generation layers. This was done in favor of simplicity and speed.
Equivariance has to do with symmetry. Molecules have symmetry because you can transform them by translating or rotating them and their properties don’t change. Equivariance is a form of inductive bias. By reinforcing it in a neural network, the model can learn easier on less data because symmetries don’t need to be learnt.
The removal of an equivariant network was confusing to some online. However, others pointed out that AF3 is still exposed to equivariance through data augmentation, a technique that has been around for quite some time. With AF3, this means that training examples were rotated or translated to help the model understand symmetries—it’s a way of squeezing more out of the training data.

Parting Thoughts
“We’re still a long way from replacing experimental methods with ML in the life sciences. That said, progress has been breakneck this decade and one day when I write one of these articles—ML will have replaced some experimental methods.”
Make no mistake, AF3 is yet another meaningful step forward for computational biology. There will be optimists and detractors. Goal posts will be moved inexorably backwards with instances of “but it still can’t do this” or “it’s still not accurate enough for this”.
I fall into the camp of “it’s still pretty early”. It’s tantalizing to think that ML will eat experimental methods—that x-ray crystallography or Cryo-EM will be subsumed by the din of humming GPUs. I don’t think even the staunchest ML advocates think we’re close to wholesale deleting experimental methods.
AF3 seems to struggle with proteins lacking co-evolutionary information (antibodies), proteins that are highly conformationally dynamic in their environments, proteins that are hard to crystallize (membrane proteins), and proteins with IDRs. Moreover, it’s yet to be determined how AF3 will generalize to novel small molecule scaffolds—which are highly relevant to drug discovery efforts.
The main takeaway I have from the AF3 odyssey and the resulting aftershocks is that we need more data. For another monumental improvement, AF3 and other similar models, will need to go beyond the PDB which has been mined to oblivion at this point. Each year of new data matters a lot, though, because it’s much more accurate and well-annotated than structures from decades ago. Plus, synthetic training regimes like using MD trajectories are incredibly exciting paths forward. All in all, AF3 is awesome and will hopefully serve as a beacon to get more outside eyeballs (and capital flow) on innovation in the life sciences.
Thanks for reading! This essay was intended for folks without a deep background in ML, like myself. If you feel I butchered something or came to a stray conclusion, feel free to DM me on Twitter (X)!
There are some other excellent blogs that dig more deeply into AF3’s performance on various tasks, including those by Eva Smorodina and Carlos Outeiral—you should read them!
I’d like to specially thank John Chodera, Charlie Harris, and Henri Palacci for their feedback and input on this piece.
excellent write-up, also great color palette