

ASCO 2025: Themes & Takeaways
After attending this year's ASCO conference, we've distilled a list of key concepts spanning checkpoint inhibition, TCR-inspired drugs, tackling treatment-resistant breast cancer, and more.


Context
Members of our team recently spent a week in Chicago for the American Society of Clinical Oncology (ASCO) 2025 conference. We attended poster sessions, walked the exhibition floor, and listened to many structured sessions. Below, we’ve catalogued some of the themes and ideas that stuck out to us. Please feel free to debate, discuss, or reach out to us if you’d like to attend our reception at a future ASCO.
Broader and Earlier: New Horizons for Immune Checkpoint Inhibitors
TCR-Inspired Therapies: A Gold Rush on Intracellular Targets
First-in-Human: mRNA-Encoded T Cell Engagers (TCEs)
Next-Gen Hormone Therapy and Beyond: The Future of Advanced Breast Cancer Management
Broader and Earlier: New Horizons for Immune Checkpoint Inhibitors
Already mainstream medicine, immune checkpoint inhibitors are poised to expand into earlier stage disease and could benefit patients when administered before and after surgery.
Throughout the last 15 years, immune checkpoint inhibitors (ICIs) have cemented themselves as a defining pillar of cancer immunotherapy treatment, beginning with the FDA approval of anti-CTLA-4 therapy in 2011 and subsequent approvals for anti-PD-1, PDL-1, and LAG-3. Specifically, ICIs function by blocking these aforementioned inhibitory cell-surface receptors, facilitating activation of T cells upon encountering target cancer cells.
To date, ICIs’ therapeutic impact has been observed across more than 20 tumor types are predominantly approved to treat late-stage, metastatic disease. These patient populations often serve as a first entry point for novel therapeutic mechanisms given faster regulatory approval pathways and a greater risk tolerance for side effects (such as the immune related adverse events often associated with ICI therapy).
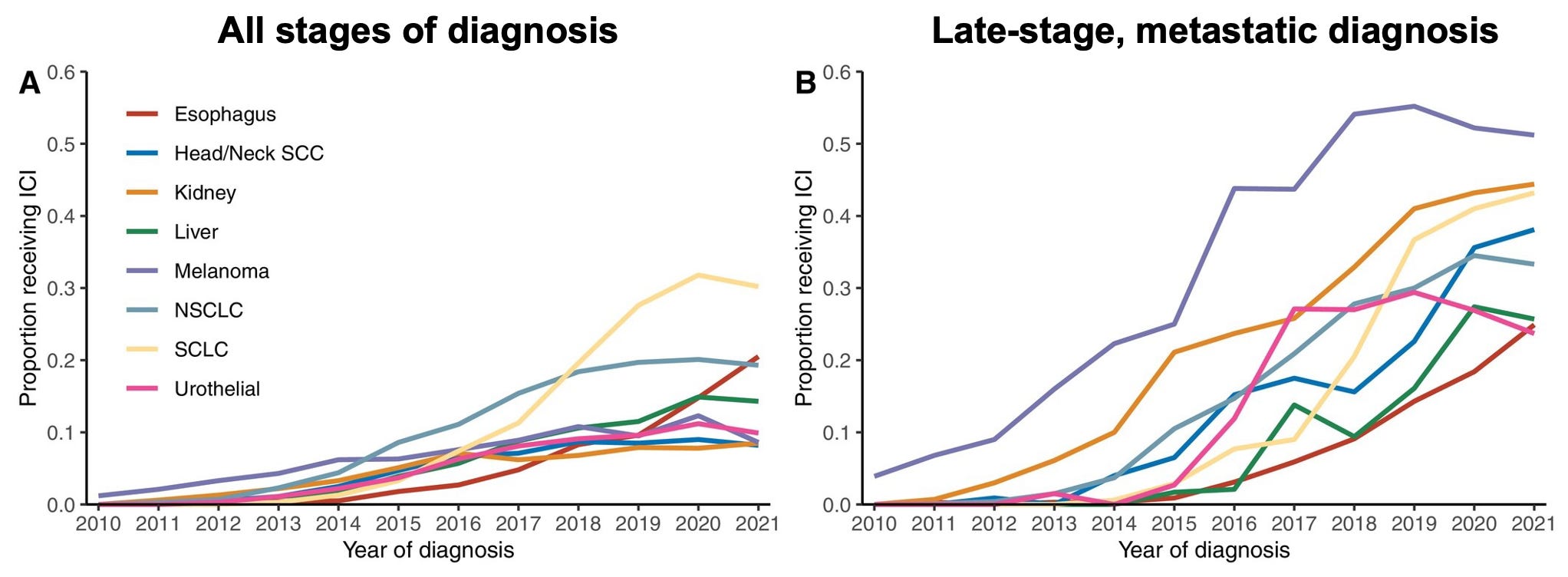

A major trend we observed at ASCO this year was the desire to expand the current clinical success of ICIs into earlier stage patient populations with new dosing and combination strategies. Here’s an overview of two clinical trial readouts we believe are good reminders of the significant life left for ICI treatments in treating new patient populations:
ATOMIC—Are ICI therapies effective in treating pre-metastatic colon cancer in combination with standard of care chemotherapy?
The current standard of care (SOC) for stage III colon cancer is adjuvant chemotherapy; however, ~30% of these patients still see tumor recurrence. ATOMIC’s Phase III trial aimed to reduce this recurrence rate via the addition of anti-PDL-1 (atezolizumab) to SOC chemo in patients with stage III, non-metastatic deficient DNA mismatch repair (dMMR) colon cancer.
dMMR tumors have inherently high mutational burdens, leading to the display of unique peptides on the cancer cell surface that selectively trigger immune cell activation and clearance of the tumor. This is the predominant reason why ICIs are so effective against these tumors—the key is already in the ignition and the ICI is there to release the brake on T cell-mediated cytotoxicity.
As hypothesized, these trial results strongly demonstrated that combination therapy of atezolizumab + FOLFOX6 chemotherapy significantly improves event free survival (EFS) compared to SOC FOLFOX6 alone—reducing the risk of recurrence and death in these patient by ~50% at 36 months post treatment.

While it should be noted that the ICI + chemotherapy combination did result in an expected increase in Grade >3 adverse events (AEs) compared to chemo alone, the improvement in EFS and remission rates most likely outweighs these concerns.
The ATOMIC trial—alongside many others attempting to do the same for other tumor types and ICI drugs—serves as a major stepping stone for expanding access to ICI treatment for patients with earlier-stage disease.
MATTERHORN—How does the timing of ICI treatment impact patient responses?
ICIs are currently approved for use with SOC chemotherapy in patients with metastatic gastric or gastroesophageal junction (G/GEJ) cancer where surgery is not possible. However, in local non-metastatic G/GEJ tumors, the clinical benefit of including ICIs in a perioperative (before and after surgery) setting is unknown.
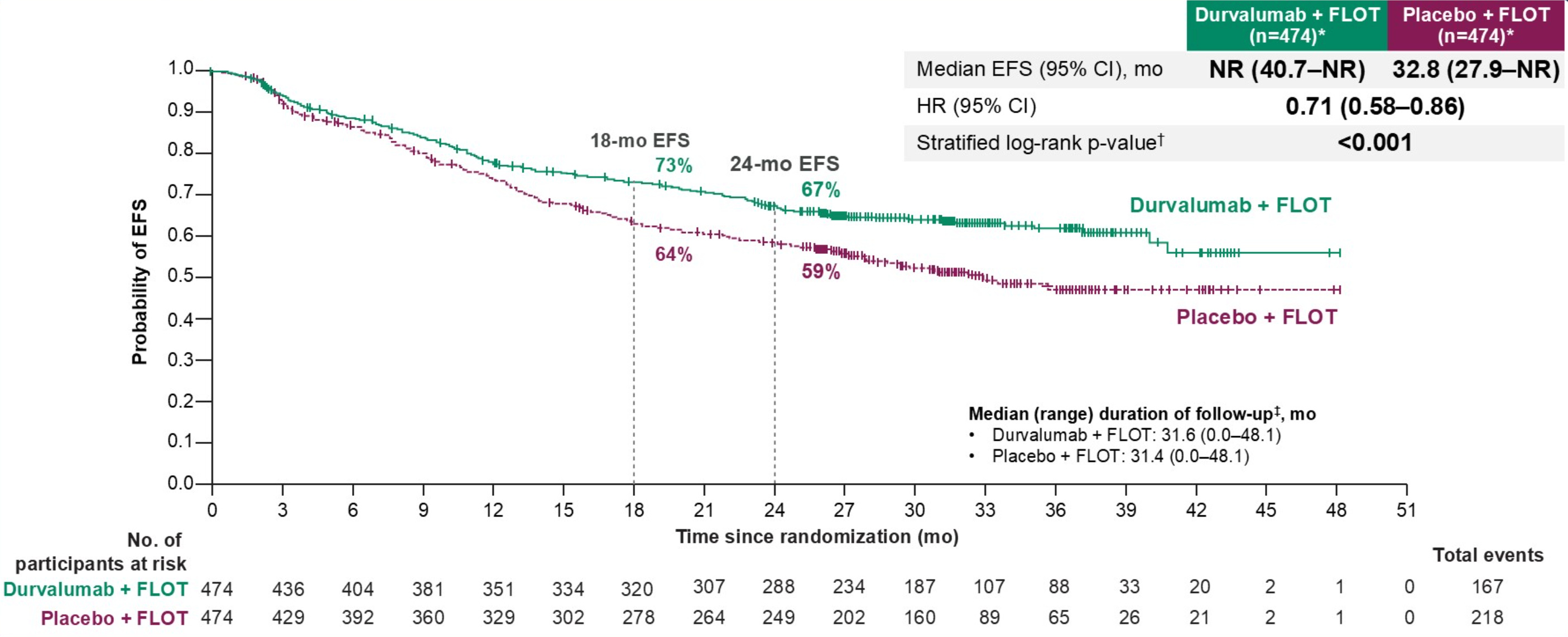
The Phase III MATTERHORN trial evaluated this perioperative strategy by testing anti-PDL-1 (durvalumab) + SOC FLOT chemotherapy in patients with resectable early-stage G/GEJ cancer compared to FLOT treatment alone.
They found perioperative durvalumab + FLOT resulted in a 67% EFS compared to 59% in FLOT alone at 24 months. This data was paired with impressive pathologic complete response rates in durvalumab + FLOT vs. FLOT alone (19% vs. 7%).
Interestingly and in contrast to ATOMIC, MATTERHORN showed almost identical rates of adverse events across treatment arms, suggesting that most of the side effects resulted from chemotherapy and/or surgery.
Here’s what we’re still wondering:
The ATOMIC trial did not include a treatment arm testing ICI treatment alone. Could the additional chemotherapy actually be dampening clinical responses to ICI if it leads to elimination of lymphocytes?
Will the increased immune-related adverse events associated with ICIs limit the reach of these therapies into even earlier stage tumors where patient risk tolerance is low? Or will drugs against newer ICI targets (LAG-3, TIGIT, TIM-3) help improve overall safety profiles with similar efficacy?
ICI therapies are still generally limited in their ability to combat immunosuppressive features of the tumor microenvironment and are subject to common resistance mechanisms. What combination therapies or broader strategies are most promising in overcoming these barriers to enhance ICI efficacy?
TCR-Inspired Therapies: A Gold Rush on Intracellular Targets
The vast majority of proteins are intracellular. This universe of potentially tumor-associated cancer targets has been off-limits to modern immunotherapies. But that may be changing.
About 26% of human protein targets are extracellular (~12% secreted proteins and ~14% cell-surface proteins) and thus accessible to common immuno-oncology treatments such as antibodies and CAR-T cells. Consequently, the remaining ~74% of targets are intracellular and generally out of reach to these modalities. Finding strategies to target these intracellular proteins—especially those uniquely expressed in cancer cells—will undoubtedly drive the next wave of precision immuno-oncology.
Across your tissues, cells continuously break down intracellular proteins into small peptides, which are then presented on the cell surface by human leukocyte antigen (HLA) receptors. Your body’s T cells are naturally equipped with unique T cell receptors (TCRs) trained to recognize specific cell surface peptides indicative of a diseased or infected state—leading to T cell activation and targeted cell death, as shown below.

Researchers have learned to mimic and engineer TCRs to target known tumor-specific peptides for selective elimination of cancer cells. While the field is still in its infancy, there are already clinical approvals that demonstrate the impact of these therapies including KIMMTRAK (a TCR-T cell engager) and Tecelra (an engineered TCR-T cell therapy).
This year’s ASCO meeting continued to show this field’s maturation with a compelling readout of Immatics’ IMA203. IMA203 is an autologous TCR-T cell therapy targeting the PRAME peptide/HLA-A*02:01 complex and currently being tested in Phase Ib clinical trials for advanced melanoma patients.
PRAME is an intracellular protein expressed across more than 50 tumor types and a member of the cancer testis antigen (CTA) family. CTAs are a group of >250 genes that are epigenetically silenced in adult tissues, but are active in ~40-50% of solid tumors—making them ideal cancer targets with interest across several preclinical and clinical TCR programs.
At ASCO, Immatics announced that IMA203 resulted in a 56% objective response rate (ORR) and a PFS of 6 months among 32 heavily pretreated, anti-PD-1 refractory metastatic melanoma patients. This is a significant increase in ORR compared to recently reported ICI treatments in similar melanoma patient populations (Source 1, Source 2), showcasing the potential for TCR based therapies.

As expected for patients who receive lymphodepletion therapy, the most common adverse event was cytopenia. Cytokine release syndrome (CRS) stayed manageable in most patients (84% Grade 1 and 2, and 11% Grade 3).
This impressive readout further validates IMA203’s Regenerative Medicine Advanced Therapy (RMAT) designation by the FDA, which enabled the swift initiation of a Phase III trial currently in recruitment for advanced or metastatic cutaneous melanoma. If all goes well, Immatics plans to submit for FDA approval in early 2027.
Here’s what still on our minds:
Current TCR-based therapies are restricted to specific HLA subtypes, thus requiring novel TCR variants to treat broader patient populations. How does the TCR field more rapidly expand their reach to HLA subtypes beyond the most common alleles (e.g., HLA-A*02:01)?
Lymphodepletion prior to injection of TCR-T cell therapies is still standard practice, yet is also responsible for serious adverse events. What strategies are being developed to mitigate side effects or eliminate lymphodepletion altogether?
Cancer cell downregulation of HLA is a common resistance mechanism for these therapies as it removes the activation signal to effector cells. How do we combat this and other resistance mechanisms?
First-in-Human: mRNA-Encoded T Cell Engagers (TCEs)
What if there were a way to skip an entire therapeutic supply chain by turning a patient’s own liver into an antibody-producing bioreactor?
Virtually all biologic drugs (e.g., antibodies) are biomanufactured at scale in gigantic, steel reaction vessels. Once purified and packaged, we ship these therapeutic molecules around the world via temperature-controlled supply chains.
Bioprocessing has improved substantially in recent decades, especially for monoclonal antibodies (mAbs). But with more complex biologics (e.g., bispecifics) gaining a clinical foothold, it’s worth revisiting the question—can we avoid industrial biomanufacturing altogether?
Scientists have long tried to leverage the body’s internal machinery to manufacture therapeutic molecules. In 1990, one group delivered mRNA directly into mouse skeletal muscle and showed evidence of in vivo protein translation. This early proof of concept has smoldered since, limited by inadequate technology.
Nowadays, we have robust infrastructure for designing and making mRNAs at scale. Lipid nanoparticles (LNPs) for mRNA delivery have been tested in millions of people owing to vaccinations against SARS-CoV-2 from BioNTech and Moderna.
Both of these companies have repurposed their mRNA-LNP technology and know-how towards making therapeutic in vivo protein expression a reality.
Moderna will launch a pivotal clinical trial this summer for mRNA-3705, an mRNA-encoded enzyme replacement therapy for a rare disease called methylmalonic acidemia (MMA). Recently at ASCO, BioNTech release early clinical data on BNT-142, the world’s first mRNA-encoded bispecific T cell engager (BiTE) for treating solid tumors expressing claudin-6 (CLDN6).
Unfortunately, BNT-142 achieved lackluster efficacy (ORR = 5%) in all tested solid tumors. But that shouldn’t be the takeaway. BioNTech’s PK/PD and safety data lend significant insight into the potential pros and cons of encoding complex, bispecific antibody formats as mRNA.
In a dose escalation format, BioNTech infused between 50 ng/kg and 100 µg/kg of a CLDN6—CD3 BiTE encoded as an mRNA-LNP to patients (n=65) with a range of advanced solid tumors. They reported BiTE PK data between 1-100 µg/kg as shown below.

There was a dose-dependent increase in serum BiTE expression in the 1-60 ng/mL range that peaked around one day after infusion with a gradual decline resulting in a median half-life around 1.5 days. This is a curious PK niche. mRNA-encoded bispecifics seem to occupy a concentration—half-life window somewhere between an IV-delivered fusion protein and bispecific-IgG antibody.
For example, BNT-142 peaked higher (ng vs. pg) and lasted longer (days vs. hours) than Blincyto (blinatumomab)—a commercially approved non-IgG BiTE composed of a fused set of scFvs. Meanwhile, BNT-142 peaked 10x lower and lived 10x shorter than Imdelltra (tarlatamab)—an analogous IgG-bispecific approved to treat small cell lung cancer (SCLC). We wonder if the in vivo translation approach may not have enough firepower to take out a solid tumor.
Generally, FDA-approved bispecifics targeting solid tumors reach peak concentration in the µg/kg range and last for weeks. Kimmtrak (tebentafusp-tebn) is a notable counterexample. Approved for metastatic uveal melanoma, Kimmtrak has a ~10 ng/mL max concentration and a half-life of 6-8 hours—an achievable set of targets for the in vivo translation strategy.
The key question remains—how does all this PK data translate to patient safety?
Cytokine release syndrome (CRS) is a systemic inflammatory response (adverse event, AE) typically brought on by immunotherapies. CRS is common AE for T cell engagers that often requires hospitalization if it exceeds Grade 2. We’re not CRS experts, but here’s our current understanding of why a mRNA-encoded TCE may help mitigate the CRS class effect.
CRS is first triggered by the intensity and synchrony of immune synapse formation. A large and fast concentration spike likely causes a strong cytokine surge. Since the mRNA-encoded version takes longer to reach its peak concentration, it may abate the initial cytokine response—regardless of the magnitude.
It reasons CRS is also linked to drug exposure. Since the mRNA-LNP approach has a shorter tail than IV-infused IgG-bsAbs, the exposure is lower too. On the other hand, you can’t “turn off” the in vivo translated version once injected, which could pose a new challenge. Compounding this is the potential for LNP-induced toxicity. BioNTech’s PD and safety data are insightful here.

At higher dose levels, BNT-142 induces a cytokine (IL-6) surge >400 pg/mL, which isn’t markedly lower than other TCEs. Each successive cycle elicits a reduced cytokine response—similar to other TCEs that require therapeutic step-ups to reduce CRS incidence. The IL-6 drop off does appear shorter than for IgG-based bsAbs, which is encouraging and could limit exposure that may trigger CRS.

Taking a look at AEs, we can see a ~20% all-grade CRS rate, which increases to ~30% at higher doses. This compares to approved IgG bispecifics which sometimes carry a ~40% all-grade CRS rate. Critically, there’s an AST/ALT signal here that is probably LNP-induced—an issue unique to this delivery modality.
Altogether—these data are encouraging, but not dispositive. The mRNA-encoded bsAb approach seems technically feasible, logistically attractive, and at least not worse than a direct protein infusion in terms of safety. Granted, this is a single, small (n=65) study that left much on the table with efficacy. We’ll need to see more.
Here’s what we’re still asking ourselves:
Can an mRNA-LNP approach thread the drug exposure needle enough to achieve meaningful efficacy while simultaneously lowering immune side effects like CRS?
Will the high local drug concentration around the liver as well as LNP-induced toxicity outweigh any ostensible safety benefits?
If both of these are true—why would this approach NOT become the future of this entire field?
Next-Gen Hormone Therapy and Beyond: The Future of Advanced Breast Cancer Management
The data revealed during ASCO plenary sessions is oftentimes practice changing. This year, advanced breast cancer took center stage—with news about emerging hormone therapies and new options for patients with treatment resistance.
Advanced breast cancer (aBC) is a scourge on society. Roughly 30,000 Americans will be diagnosed with aBC this year and another 200,000 are currently living with the disease. About a quarter of patients diagnosed with early stage breast cancer (BC) will unfortunately progress to metastatic disease, underscoring the need for better treatment options.
Over the past twenty years, however, the five-year survival rate for patients diagnosed with aBC has expanded monotonically from 25% to 34%—a final figure which doesn’t capture the efficacies of today’s best treatments.
Oncologists typically divide BC into four molecular subtypes defined by hormone receptor (HR) status and HER2 protein expression levels. Representing ~70% of annual cases, HR(+)/HER2(-) tumors are the most common subtype. Given the large unmet need, many new therapies focus on this subpopulation.
HR(+) tumors use the hormone estrogen as fuel to dysfunctionally upregulate key cell cycle proteins—resulting in uncontrolled cell growth. Oncologists can counter this by disrupting estrogen receptor (ER) signaling. Modern hormone therapy includes drugs like selective ER modulators (SERMs), selective ER degraders (SERDs), and aromatase inhibitors (AIs) that do the following:
SERMs competitively bind tumor-expressed ERs, blunting estrogen signaling before it reaches the nucleus.
SERDs antagonize ER activity and trigger the ubiquitination and subsequent degradation of ER proteins.
AIs halt the conversion of androgen to estrogen, thereby lowering the serum concentration of the key signaling molecule responsible for cancer growth.
Recently, a wave of FDA-approved precision therapies has earned its place alongside hormone therapy. Since 2015, several CDK4/6 inhibitors have entered into the standard of care (SOC) for first line (1L) management of HR(+) aBC.
These drugs (e.g., palbociclib, ribociclib, abemaciclib) all inhibit the activity of key cyclin dependent kinases (CDK4/6) responsible for regulating the cell cycle. Since CDK4/6 overlap with a node in the ER signaling pathway (cyclin D1), scientists reasoned there would be combination synergy with hormone therapy.
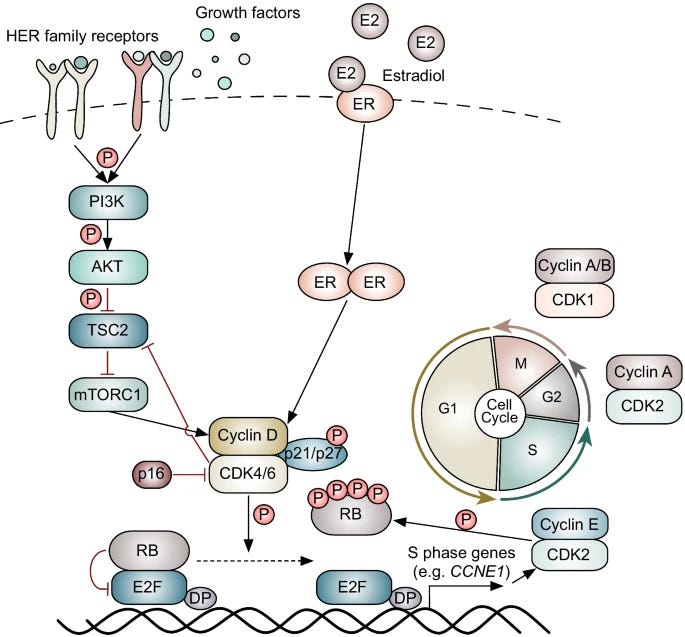
In combination with hormone therapy, CDK4/6 inhibitors have shown to improve progression free survival (PFS) and overall survival (OS) across multiple Phase 3 clinical trials, as shown below.

Unfortunately, advanced HR(+) tumors often adapt to hormone therapy. Common resistance mechanisms can be ER-dependent (ESR1 mutations) or ER-independent (GF-RTK alterations, MAPK alterations, or PI3K-AKT alterations). These genomic drivers of resistance have become the targets of a flurry of clinical development activity.
Moreover, data shown at ASCO suggest that some of these resistance mutations also have prognostic and therapeutic implications that the field will continue wrestling with for some time.
Tackling ER-Dependent Resistance with Next-Generation SERDs
The gene ESR1 encodes human ERs. Only 1% of patients harbor ESR1 mutations when diagnosed with aBC, though around 40% will develop ESR1 mutations following 1L treatment with hormone therapy.
Typically, ESR1 mutations interfere with the binding of some SERMs or SERDs, causing them to lose effectiveness. ESR1 mutations may also lock ERs in a conformation that promotes constant (constitutive) signaling even in the absence of the estrogen ligand—causing dose-dependent resistance to AIs.
Recent clinical trials, including those that read out at ASCO, have helped clarify how we should treat patients with ESR1-mediated resistance. Elacestrant was the first next-gen, oral SERD to show increased activity over SOC in patients presenting with ESR1 mutations at the time of disease progression.

The 2024 EMBER-3 trial similarly showed that imlunestrant has durable activity in ESR1-mutant disease, and increasingly so when combined with a CDK4/6 inhibitor. It’s key to note that the imlunestrant-abemaciclib doublet regimen also caused substantially more toxicity, as outlined below.

This year at ASCO, data from the SERENA-6 study of camizestrant demonstrated that patients who switched from an AI + CDK4/6i to camizestrant + CDK4/6i had a median PFS expansion from ~9 months to ~16 months. This effect was especially pronounced for patients with clonal ESR1 mutations.
Interestingly, SERENA-6 randomized patients following the detection of ESR1 mutations on a circulating tumor DNA (ctDNA) test rather when the disease progressed radiographically. We know that modern ctDNA tests can find molecular recurrence six months prior to radiographic progression, depending on the cancer.
The previous SERENA-2 trial showed that a >50% reduction in ctDNA ESR1 allele frequency was associated with a near-tripling of PFS, indicating prognostic utility for ctDNA assays at the least.
Cautiously, the boost in PFS could stem from lead time bias. It’s likely that guiding adjuvant treatment using ctDNA will prove useful, though the balance between the certainty of toxicity (especially in doublet regimens) versus PFS extension isn’t clear yet. We’d want to see OS or second progression free survival (PFS2) data first.
Everyone was clamoring to hear the news from VERITAC-2—the first ever Phase III evaluation of an ER proteolysis targeting chimeric (PROTAC) degrader (vepdegestrant). Standard SERDs destabilize ERs, leading to their degradation. Meanwhile, PROTACs actively recruit proteosomal degradation machinery and work catalytically—two potential pharmacological advantages.
The data showed that vepdegestrant monotherapy conferred a ~3 month PFS increase when compared to fulvestrant alone in the ESR1 mutant subpopulation—a figure that Wall Street didn’t seem to like. Perhaps this was because the drug didn’t add value in the all-comer setting or because the PFS improvement was seen as too small.
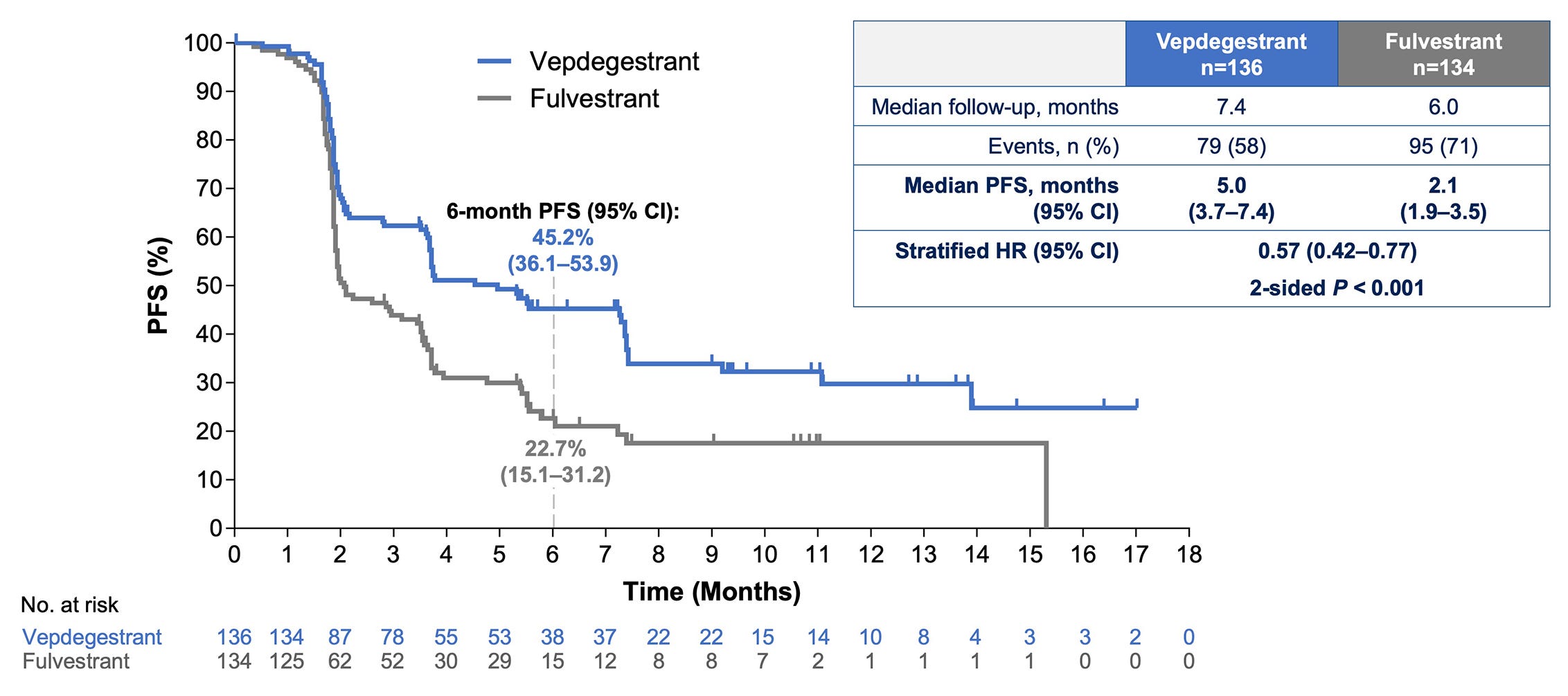
There could still be merit here. Provided OS is similar to fulvestrant, an oral monotherapy alternative like vepdegestrant could have a place in the market. However, monotherapy in the 2L setting seems to be becoming increasingly rare.
With its safety profile, vepdegestrant could allow patients to avoid beginning chemotherapy for longer. We’d argue that a 3 month PFS improvement isn’t something to scoff at. Marschner et al. showed in 2020 that disease progression causes an immediate, substantial decrease in quality of life—every month matters.
Overcoming ER-Independent Resistance with Novel Agents
Therapeutic pressure from hormone therapy pushes some tumors to compensate through alternative pathways like GF-RTK, MAPK, and PI3K-AKT—the latter being the most common. During ASCO, the speakers discussed multiple, ongoing efforts to drug the PI3K-AKT pathway.
Five years ago, the SOLAR-1 study showed that adding a PI3Kα-specific inhibitor (alpelisib) to SOC fulvestrant meaningfully increased PFS in the PI3K-mutant subpopulation—with roughly equal robustness across common PI3K variants.
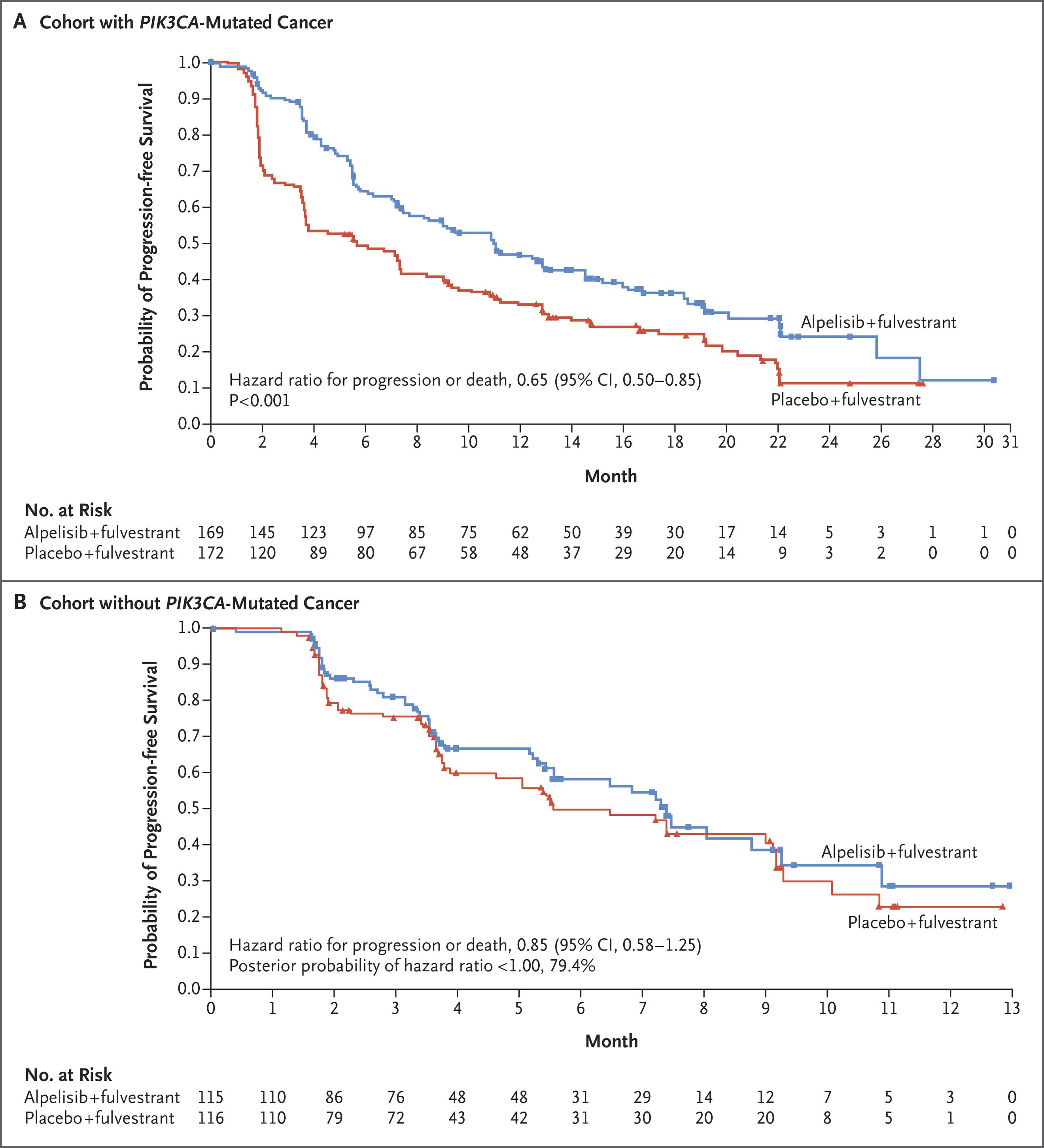
SOLAR-1 set the stage for several newer studies, including the INAVO-120 Phase III trial of inavolisib—an isoform selective inhibitor and degrader of PI3Kα. Combined with CDK4/6i and fulvestrant, inavolisib more than doubled PFS in the PIK3K-mutant population. Critically, INAVO-120 also showed an OS benefit—the first time this has been demonstrated with the class. We’ll also note that PI3K status was determined using ctDNA, which poses yet another risk for capturing lead time bias. We’re looking forward to seeing the forthcoming wave of allosteric, mutant-resistant PI3K inhibitors.
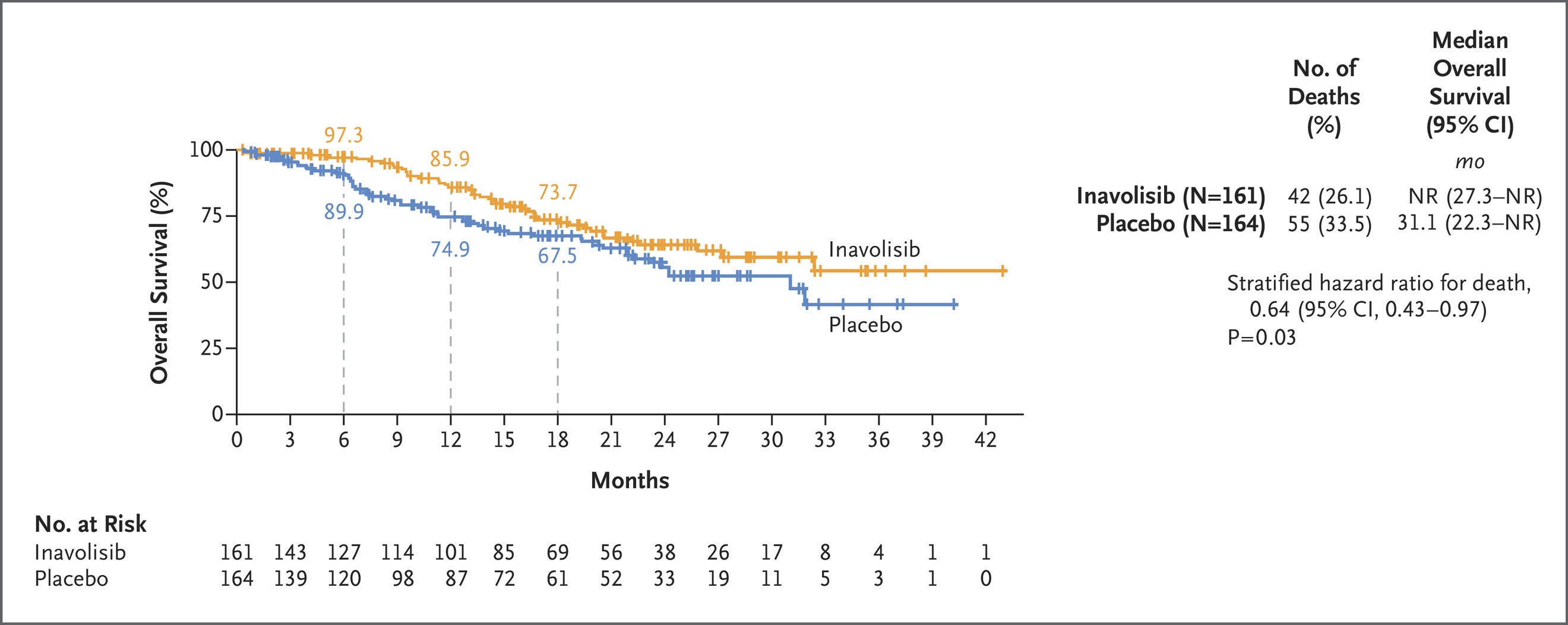
PI3Ki and CDK4/6i have sparsely been combined due to toxicity overlap. Instances of hyperglycemia, diarrhea, rash, and stomatitis were all higher in the inavolisib group. Hyperglycemia was particularly an issue in patients who had a high BMI or were otherwise pre-diabetic. Another study further characterized hyperglycemia in these at-risk patients, showing that treatment with inavolisib required additional anti-diabetic therapy and occasional dose interruption.
On the AKT front, the 2023 CAPITELLO-291 study demonstrated that capivasertib—a pan-isoform AKT inhibitor—provided a more pronounced benefit in patients whose tumors harbored an AKT pathway alteration. The more recent FINER study echoed these results with ipatasertib—another pan-isoform inhibitor. This trial showed both a PFS and OS benefit in the ITT and AKT-altered cohorts. ctDNA was used for randomization here as well and the OS data was only at 50% maturity, however.

Here’s what we’re still musing over:
ctDNA is inevitable, but how can we use it most effectively? Will it become a surrogate endpoint for solid tumors? How do we structure trials in a way to avoid capturing any lead time bias?
Will emerging isoform or mutant-selective allosteric inhibitors for PI3K or AKT add a material boost to efficacy or safety?
Will attacking CDK2/CCNE1 upregulation allow for more complete cell cycle arrest when combined with CDK4/6 inhibition?
If doublet or triplet regimens become the norm, how will we think about AEs in both curative and palliative-intent scenarios?
How should we think about cross-reactivity and sensitization given the overlap in resistance pathways?