

Enlicitide: A Curious(er) Macrocycle
Macrocyclic compounds lend themselves to interesting pharmacology. Merck's emerging PCSK9 inhibitor for "bad" cholesterol is no different. Will it retain its pole position or will it get leapfrogged?

Background
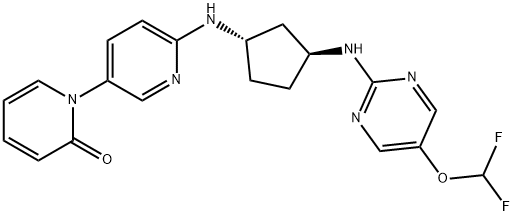
Earlier this week, Merck hinted at positive interim results from several ongoing Phase 3 clinical trials evaluating the efficacy of enlicitide, the company’s oral PCSK9 inhibitor. As some have pointed out on Twitter, enlicitide is an enormous macrocyclic compound with a 37-member ring.
I love writing about macrocycles. These beautiful monstrosities are challenging to discover, difficult to optimize, and tedious to scale. Therefore, companies that choose to develop macrocycles usually have a very compelling reason.

In another post, I wrote about how macrocycles are uniquely suited to modulate intra-cellular protein-protein interactions (PPIs)—a pharmacological niche that’s usually off limits to both traditional small molecules and larger biologics. I focused on REC-6236, a fascinating glue-like macrocycle that disrupts RAS signaling in pancreatic cancer.
Now I want to dig into enlicitide to explore all its quirks, features, and its reason for existence in the first place—starting with a bit of biology.
PCSK9 and Bad Cholesterol
You’ve probably heard of bad cholesterol, otherwise known as low-density lipoprotein (LDL) cholesterol. As with most things, it’s the dose that makes the poison. LDL cholesterol isn’t inherently bad—these molecules help give cell membranes their structures.
However, LDL does have a tendency to oxidize inside arterial walls, triggering an inflammatory response that results in plaque formation (atherosclerosis). The blocking and narrowing of coronary arteries significantly increases one’s risk for major cardiovascular episodes like heart attack or stroke.
The liver is the central regulator of LDL in our bodies. The majority of LDL is produced in and excreted by the liver, where it’s free to travel throughout the circulatory system.
If LDL is gets too high, hepatocytes upregulate the expression of LDL receptors (LDLRs), which grab soluble LDL molecules and pull them back into the liver—decreasing serum LDL.
When LDL is too low, PCSK9 gets involved. This 692-residue protein is also synthesized in the liver. Once in the circulatory system, PCSK9 also binds to the surface of LDLR, triggering receptor-mediated endocytosis.
Inside the acidic pH of an endosome, the PCSK9-LDLR affinity intensifies and the complex is marked for degradation inside lysosomes. With fewer surface-exposed LDLRs, the liver cannot pull LDL inside as rapidly—increasing serum LDL.
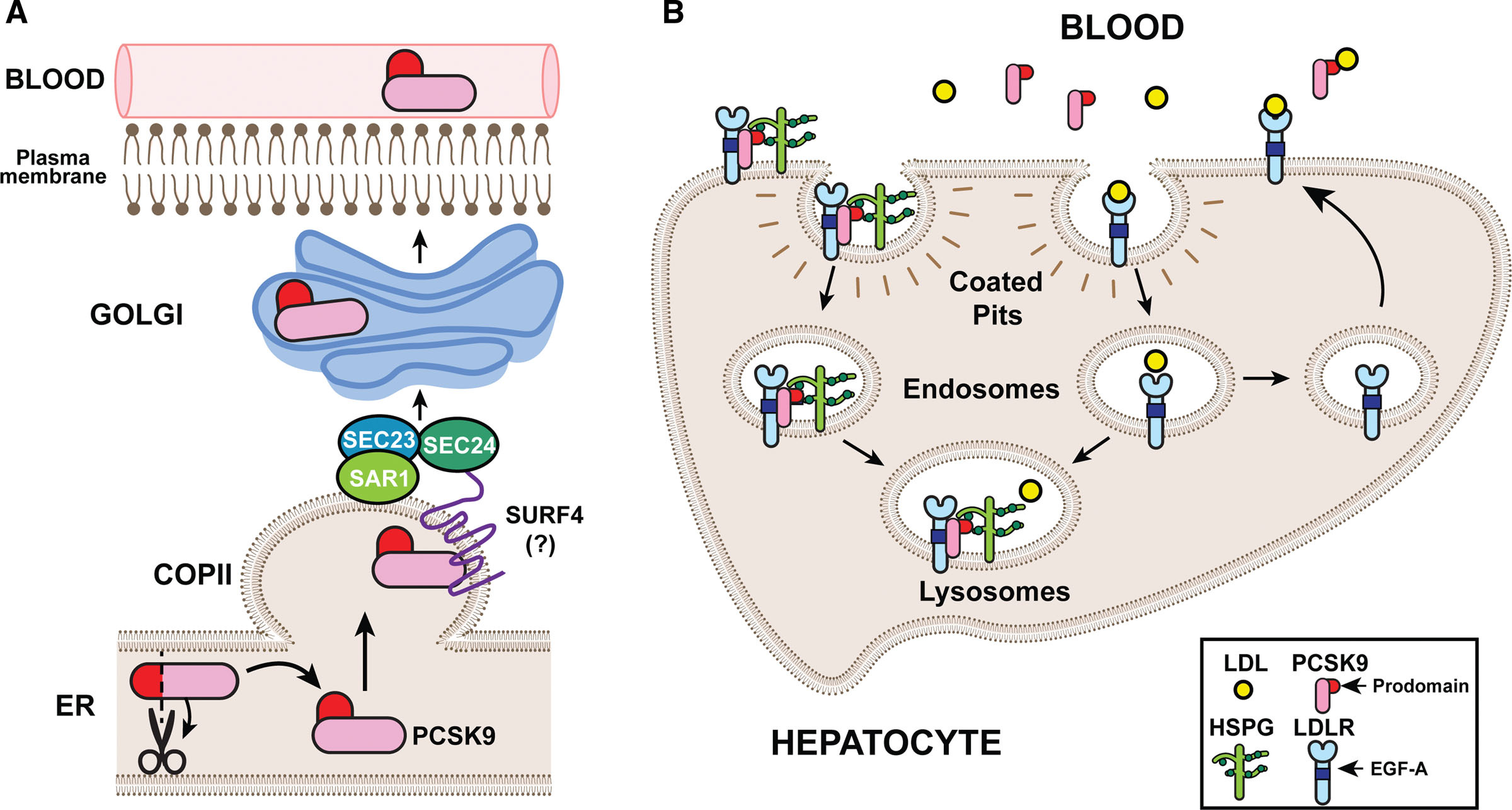
Given PCSK9’s central role in regulating LDL, it should come as no surprise that there’s a strong genetic association between PCSK9 variants, LDL levels, and cardiovascular episodes.
Genetic Target Validation
Drug targets with statistical or functional genetics support are ~2.6X more likely to succeed in the clinic. Widespread sequencing and increasing access to structured, clinical data has resulted in thousands of causal genotype—phenotype linkages. Virtually all of these have been confined to diseases with simpler, Mendelian modes of inheritance.
PCSK9 is a unique case for many reasons. Firstly, LDL cholesterol levels and major cardiovascular events are complex (non-Mendelian) phenotypes. Secondly, PCSK9 is actually the fastest ever example of a genetic target translating into an approved medicine at just 12 years.
In 2003, scientists discovered missense mutations in PCSK9 that seemed to cause hypercholesterolemia—a genetic predisposition to having chronically high levels of LDL cholesterol. Just two years later, another group showed that loss-of-function (LoF) mutations in PCSK9 led to a 28% reduction in serum LDL as well as an 88% reduction in major coronary artery events over a 15 year period. Critically, these LoF alleles didn’t seem to cause adverse effects otherwise.
Taken together—these data suggested that even complete PCSK9 ablation would be both safe and potentially desirable as a therapeutic means to rescue patients with high LDL.
The First Wave of PCSK9 Therapies
Since the 1980’s, statins have been the standard of care for addressing high LDL. These compounds inhibit HMG-CoA reductase in the liver, an enzyme responsible for LDL synthesis. Statins don’t work for a lot of people, though—whether that be because of high baseline LDL levels, diet and exercise, or treatment discontinuation.
The FDA approved Praluent (alirocumab) in 2015, an antibody inhibitor of PCSK9. The second antibody called Repatha (evolocumab) was approved a few months later. Both of these antibodies bind circulating PCSK9, hindering its ability to downregulate LDLR expression—thus lowering serum LDL levels.
In pivotal trials (1, 2), both molecules achieved placebo-adjusted serum LDL reductions of ~60% from baseline when combined with statin therapy. This level of LDL reduction is clinically meaningful as it resulted in a ~15% reduced risk for cardiovascular death. Sales of both Repatha and Praluent totaled around $3 billion in 2024.

I’d be remiss to not also mention Leqvio (inclisiran), an siRNA inhibitor of PCSK9. Leqvio’s advantage is that it requires only bi-annual subcutaneous dosing to disrupt PCSK9 translation—achieving a >50% placebo-adjusted reduction in serum LDL levels. Antibodies typically require bi-weekly dosing by comparison.
A Need for New Therapies
Even with three current-generation options, the situation on the ground isn’t ideal.
Roughly half of patients discontinue PCSK9 inhibitors after just six months—the primary reason being high out-of-pocket (OOP) costs. This is a multifaceted issue that spans high copayments, deductible structures, prior authorizations, and more.
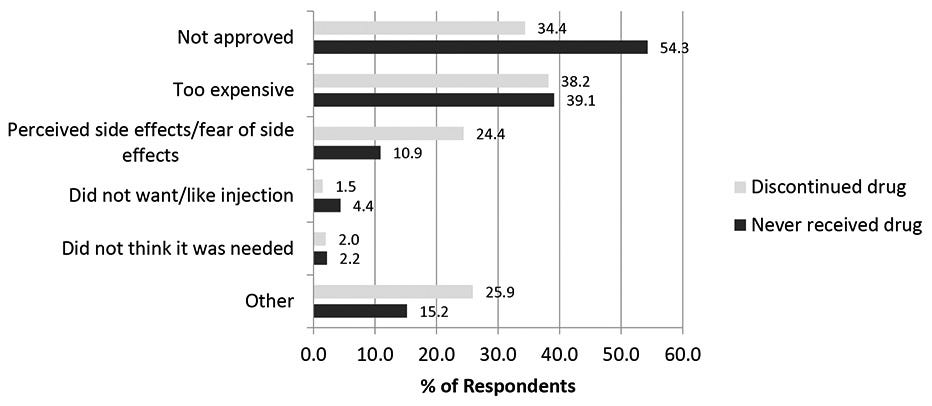
That most PCSK9 inhibitors are biologics is one contributing factor. Produced in living cell cultures, purified through multiple steps, maintained under strict temperature control, and administered via syringe—antibodies have a high cost-of-goods (COGS), some portion of which trickles down to patients.
Even non-generic small molecules, such as Lipitor (atorvastatin), cost about $150/month OOP—below the ~$200 threshold where there is substantial price sensitivity.
Sure—the fact that small molecules are oral definitely helps with patient choice. I don’t love needles either. But I found that only ~1.5% of patients discontinue their PCSK9 regimen because of the route of administration. Cost seems to be the main target product profile (TPP) niche to poke at.
If you want to read more about TPPs—here’s a shameless plug for our most recent article.

Merck’s Shot on Goal with Enlicitide
Merck chronicled the discovery, development, and early clinical testing of enlicitide in a 2023 journal article. Unsurprisingly, they cite PCSK9’s lack of a canonical active site as the primary motivator behind the macrocycle campaign—along with the possible oral route of administration.
The company’s scientists leveraged an mRNA-display platform to screen for potential macrocyclic binders. Experimental crystal structures from enlicitide analogues suggest that the macrocycle binds a flat surface in PCSK9’s non-active catalytic domain, preventing the protein from binding LDLR.
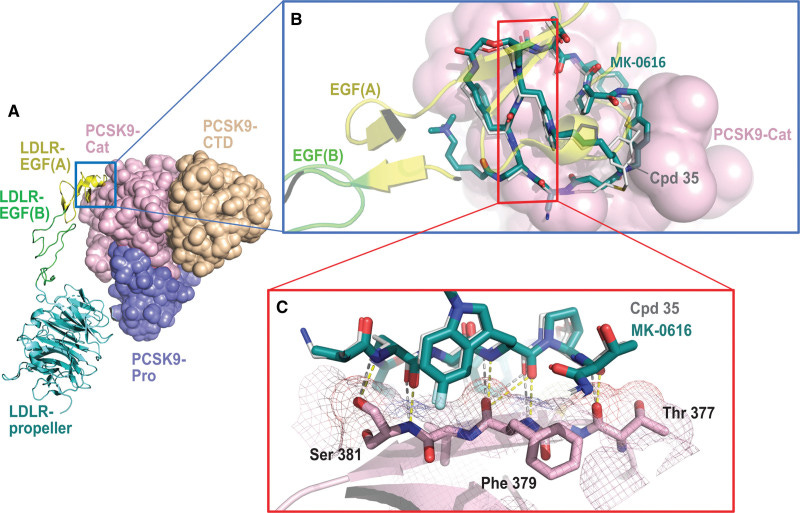
I found the first-in-human PK data and commentary rather interesting. Macrocycles, and other large (beyond-rule-of-five) compounds, typically have poor oral bioavailability and cell permeability. That is indeed the case here. The group had to jump through several hoops to get enlicitide to do its job in humans.
Patients had to be in a fasted state before taking the study drug. Moreover, enlicitide had to be co-administered with a gut permeability enhancer (sodium caprate) to get the molecule to squeeze through intestinal gap junctions and into circulation.
After all this, they showed a ~60% (non-placebo-adjusted) decrease in serum LDL. Here’s another curious thing—the idea that a lack of cell permeability could actually be a feature and not a bug.
The group hypothesized that since enlicitide cannot enter cells, the dose level can be titrated quite high as it theoretically shouldn’t interact with intracellular targets. This reason was cited as a potential factor behind the relatively clean safety profile in the Phase 1 study.
In Merck’s Phase 2 study, they mostly replicated the previous result—showing a ~55% placebo-adjusted serum LDL level reduction at the highest enlicitide dose (30 mg QD) with an adverse effect profile very similar to placebo. As for the full set of Phase IIIs, we’ll have to see.
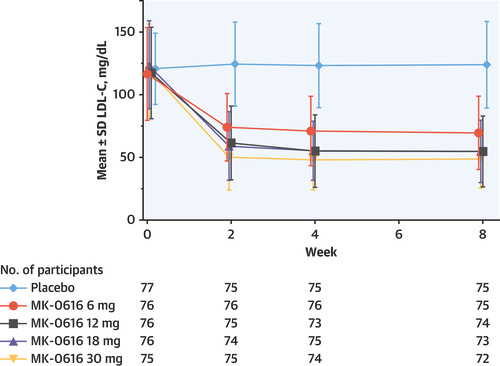
Overall, enlicitide seems like a viable and potentially first-in-class oral inhibitor of PCSK9. Unlike most macrocycles, Merck isn’t targeting an intracellular PPI. Instead, the primary motivators for this cyclic compound seem to be the oral route of delivery, the potentially lower cost to patients—and likely their belief that only a macrocycle would have enough total polar surface area (TPSA) to disrupt the binding of PCSK9 and LDLR.
There are others who seem to believe slightly differently, though.
AstraZeneca’s Dark Horse—AZD0780
Right behind enlicitide is AZD0780, AstraZeneca’s oral PCSK9 inhibitor which recently completed a Phase IIb trial. Unlike enlicitide—AZD0780 is a traditional small molecule acquired from Dogma Therapeutics way back in 2020. Since the acquisition, AstraZeneca has thoroughly nuked Dogma’s website, so I couldn’t find out much about this molecule.
My understanding is that Dogma launched a thorough structural discovery campaign to elucidate a novel, cryptic pocket on PCSK9. The company then developed picomolar binders to this pocket which could remodel the catalytic surface, thereby disrupting complex formation with LDLR.
While the structures don’t match, I was able to find another contemporary small molecule program by an Australian biotech called Nyrada—who generously published a more comprehensive explainer on their approach.
Nyrada screened compounds against PCSK9, finding that Nilotinib was a weak inhibitor that bound to a cryptic pocket proximal to the canonical LDLR binding interface. The group optimized this molecule to improve its potency. It seems that this program was placed on the back burner while the company prioritizes other lead programs.

Anyway—back to AstraZeneca.
The Phase IIb study seems competitive with enlicitide. AZD0780 conferred a ~50% placebo-adjusted decrease in serum LDL in the 30 mg QD range with a slightly better AE profile than Merck’s drug—though cross-trial comparisons are full of pitfalls. The company was sure to mention in their press release that this efficacy was achieved without fasting—and we all know who that comment was aimed at (wink, wink).
Wrapping Up
Enlicitide is yet another curious macrocycle. It seems likely that it will become one of, if not the first, approved oral inhibitor of PCSK9. Future doctors will have at their disposal a set of biologic PCSK9 inhibitors, excellent oral alternatives, an RNA-based medicine, and potentially even an in vivo gene editing solution to permanently ablate PCSK9 in the liver.
On the oral front—I’m leaning slightly more towards AstraZeneca’s molecule for a few reasons that I would love to debate for those paying closer attention to this than I am:
Both these drugs are oral, but AstraZeneca’s drug doesn’t require fasting.
AstraZeneca’s drug is likely cheaper to produce, giving them better pricing and scaling leverage.
That enlicitide can’t get into other cells doesn’t seem to be translating into vastly better tolerability.
Combined with statins, the efficacy seems about the same.
Overall—I’m happy my fellow needle-haters will have more options. What will the next curious macrocycle be?