

NeurIPS 2023 Roundup: Generative Molecular Design
Similar to therapeutic proteins, NeurIPS 2023 was packed with engaged discussions around the opportunities (and pitfalls!) of generative chemistry models. This is Part 2 of our 3-Part roundup series.

Intro
Welcome to Part Two of a three part series recapping some of my ML x Bio highlights from NeurIPS 2023. This summary isn’t exhaustive. Readers are encouraged to comment about other talks they sat in or contact me to point out anything I might’ve misunderstood.
Part Two—Generative Chemistry
Part Three—Drug-Target Interaction (DTI) Prediction
Overview
These talks took me right back to one of my favorite 2023 review articles about structure-based generative modeling. The review was upbeat, but also a harbinger for the limitations of current generative approaches.
I’m pretty optimistic myself, though I still belief generative protein design is more tractable currently than generative chemistry. There are two main reasons I feel this way—both having to do with data:
Data Volume—We can design and express enormous batches of protein variants, which isn’t yet feasible with small molecules (some are working on this). Attributing function to variant identity has become much easier with high-throughput sequencing. Though DELs can do something similar for small molecules, the bottom line is that the volume and veracity of protein data is higher.
Data Distribution—The waves of evolution have shaved many rough edges off of the structure-function manifold for proteins. In my last article, this was one reason why many have turned to inverse folding methods for designing protein binders—optimizations in sequence-space are tricky. Chemical structure-activity manifolds are laden with activity cliffs, where small changes in structure create massive changes in chemical properties. Rocky optimization surfaces are harder to traverse than smooth ones.
This borders on being a bit too philosophical—if anyone disagrees or wants to chat, I’m all ears. As far as the NeurIPS talks were concerned, here were my main takeaways:
Multi-parameter optimization is needed across proteins and small molecules.
Whether its thermostability for an antibody or permeability for an inhibitor, co-optimizing candidates for multiple endpoints is critical. Oftentimes these can pull in different directions. It’s a tall order, but one that’s necessary for making a good drug, not just a good molecule.
Some inductive bias isn’t bad.
Chemists don’t assemble molecules atom by atom, they build structures from fragments. Choosing better (read: reflective of real-world) representations of ligands can be helpful during generative modeling.
Benchmarks can be sobering, but they’re a necessary pill to take.
It’s human nature to dream of monotonic improvement. The reality is that developing rigorous benchmarks for model performance is just as crucial as model building. Without a true north, it’s tough to make progress. This point is one that Pat Walters makes often.
Let’s get to it.
Generative AI for Structure-Based Drug Design
Ron Dror (Stanford) presented his lab’s work on fragment-based generative chemistry models. These included an equivariant graph network (FRAME) and what appeared to be a related, forthcoming diffusion model.
We kicked the session off discussing why binding affinity is table stakes. Ideally, generative models produce molecules with high target selectivity, synthesizability, and satisfactory ADME properties. Dror remarked that ML models often produce technically valid, but cringey ligands. They don’t break physical laws, but a medicinal chemist would wince at the structure (e.g., it has a vanishingly-low half-life).
Throughout the talk, the conceptual through-line was that “chemistry thinks in terms of fragments”.
Published last November, fragment-based molecular expansion (FRAME) doesn’t generate a ligand de novo. FRAME grows a full ligand outwards from a core fragment that’s bound to a target protein’s binding pocket. The initial core usually is small, weakly binding, with just meh chemical properties.
FRAME is an equivariant graph network armed with a vocabulary of chemical fragments and two choices to make—(1) which direction should the next fragment go and (2) what fragment (and geometry) is best to use? The model iteratively scores each trajectory along a continuum of parameters (e.g., affinity, developability), as outlined below.

To support FRAME’s validity, Dror showed the model’s ability to generate a candidate ligand with superior affinity, selectivity, and synthesizability to an iteratively docked ligand. Notably, the candidate nearly approached the performance of an ostensibly held-out clinical molecule from Roche. The Maduke Lab at Stanford created experimental data showing a FRAME candidate outperformed an established reference binder of CLC-Ka on the basis of affinity. I wish I could’ve seen how this molecule compared on experimentally-determined ADME properties.
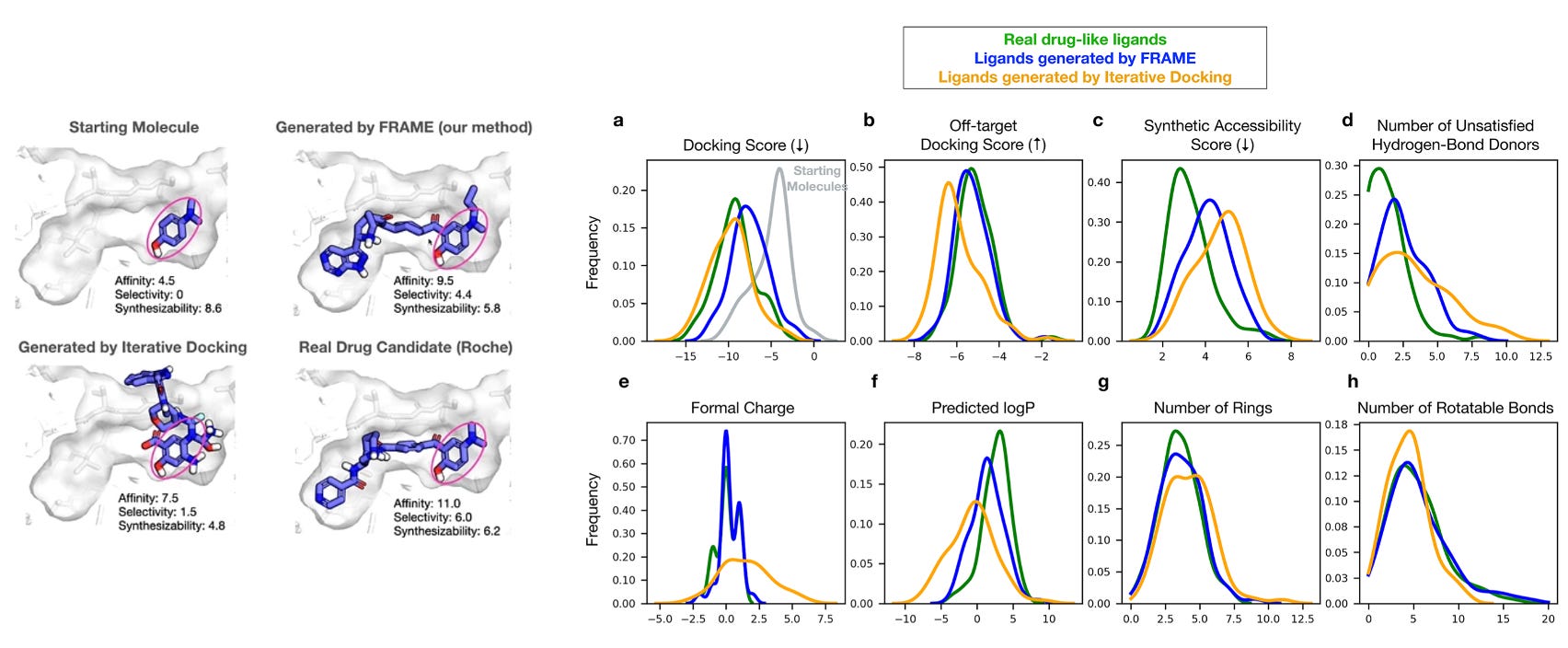
FRAME was tested on a carefully pruned subset of holo structures (n=4,200) in the PDB. This subset excludes weird binders (e.g., lipids, nucleotides). The remaining bound ligands were processed into active fragment trajectories. My question would be around target diversity—there are only a few hundred unique holo structure in the PDB with <15% sequence identity.
The model was tested on candidate and FDA approved ligands. There are roughly 2,700 unique targets with approved small molecules. How well does FRAME’s test set compare to this target diversity? Some other questions I’m eager to understand:
Could FRAME be expanded to work without pre-specifying the protein binding pocket (vis-à-vis blind docking)?
How does FRAME compare to iterative docking in terms of compute/speed?
Possibly naïve—how does FRAME calculate selectivity? Is it calculating affinity towards the set of anti-targets?
My favorite audience question was, “When does the model know when to stop building?” In industry, apparently it’s common to set a target molecular weight—which is where FRAME stops also (for now).
The talk concluded with the question—what if I don’t have a core fragment? They proposed using a diffusion architecture similarly tasked with selecting, orienting, and attaching chemical fragments. This isn’t built yet (I don’t think)—but I’m excited to see it!
As an aside—recent Dror Lab alumni published a pre-print in December on ATOM-I, a groundbreaking RNA foundation model of structure and function. It apparently uses a similar architecture to FRAME.
The Discovery of Binding Modes Requires Rethinking Docking Generalization
It was great to hear Gabriele Corso give an update on DiffDock—a diffusion model for blind docking that swept the community by storm in 2022. Blind docking refers to docking a ligand over a protein’s entire surface without knowledge of the target pocket. This method is useful for reverse screening or investigating the mechanisms underlying toxicity. To introduce how DiffDock is evolving, Gabriele cast blind docking as an NP-hard problem.
“It is easier to check that a [binding] pose is good than to generate a good pose.”
Said differently, it’s computationally intractable to guess-and-check all possible ligand binding sites and poses for a given protein’s surface. It’s relatively trivial to calculate the affinity of a given pose. This insight spawned the key conceptual advance in this DiffDock update—Confidence Bootstrapping.
According to Corso, confidence bootstrapping is a self-training method that enables DiffDock to partially overcome the data sparsity problem that plagues other ML-based docking methods. I’ll explain the problem first, then I’ll walk through the authors’ solution.
There’s scant structural data of protein-ligand complexes in the public domain (e.g., in the PDB) for training models.
Even protein sequences with <30% sequence identity (the standard cut-off) have highly structurally similar binding pockets—causing ML models not to generalize very well.
The group opted for confidence bootstrapping as it helps model builders contend with data sparse regimes and it reduces the NP-complexity of the blind docking problem. This training method involves combining a diffusion model with a confidence scoring model. I’ll walk through how I think this works:

A reverse diffusion process begins to ‘roll-out’ potential ligand poses.
Poses with high confidence are fed back in as training data, helping to guide the early stages of the diffusion process.
Maybe I’m wrong, but this seems like a method to constrain a diffusion process towards more potent binders using synthetic data (the bootstrapped, high confidence poses). I wish I understood better what confidence actually means here—is there a virtual screening-like scoring process happening under the hood? How can you prevent errant scoring from corrupting the training data?
I laud the authors for engineering a new blind docking benchmark called DockGen to assess the bootstrapped version of DiffDock. DockGen goes beyond sequence similarity cutoffs to include structural clustering around protein binding domains. Using the new training method, the team fine-tuned DiffDock on eight protein families and compared it to the previous version of DiffDock. They also compare to search-based methods, as shown in the charts below.

It’s important to point out in the left chart that without some early high-confidence poses, bootstrapping is ineffective at improving performance (e.g., clusters 4 and 5). I’m also still unsure whether DiffDock and search-based methods are being compared fairly—a point brought up in another paper. Industry-standard docking software was built to dock into a pre-specified pocket, unlike DiffDock. Please someone correct me.
PoseCheck: How Good Are Generated Poses From 3D SBDD Models?
Charlie Harris gave a well-received and rigorous overview of his group’s work on PoseCheck—a comprehensive benchmark suite for structure-based generative algorithms. My overarching takeaway is that generative models are still poor at producing viable ligand poses and that common post-processing steps mask this issue.
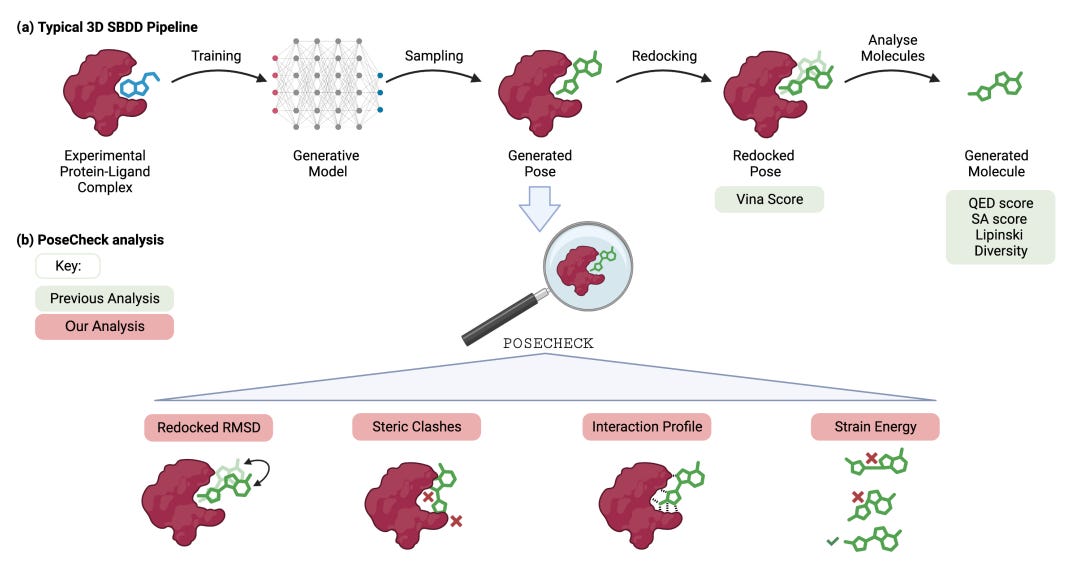
PoseCheck is a critical tool to assess the generative process. Otherwise, if we’re going to redock every time, what are we actually doing? The new benchmark suite evaluates generated poses on four interaction-based criteria that ask the following questions:
Redocked RMSD—How different is the generated pose to the one that minimizes the energy function of the bound complex?
Steric Clashes—Do models produce poses forcing non-bonding atoms too close together?
Interaction Profiles—Do generated poses have enough key interactions for tight binding?
Strain Energy—Are models creating poses that twist ligands up into energetically unfavorable states?
The headline is that many generative algorithms fail on all of these metrics. That said, I think it’s critical to walk through the data and keep in mind that this isn’t a knock on the space. We need rigorous benchmarks to ensure we’re actually moving in the right direction. Onwards.
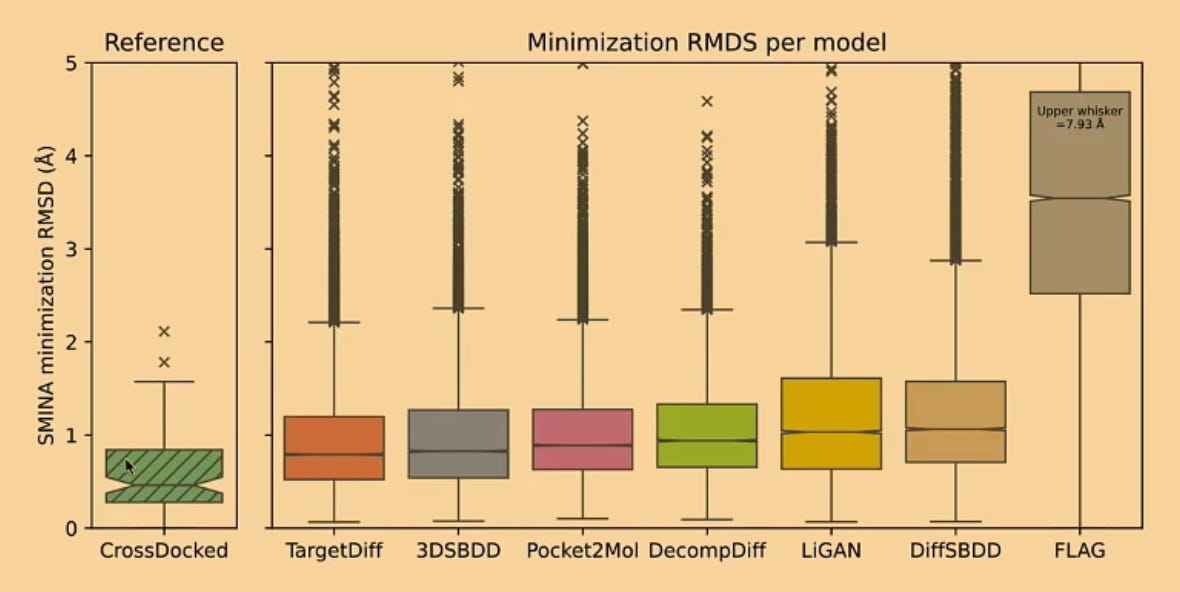



For those tested, it’s clear that generative models tend to produce poses that aren’t in an energy minimum, that have a high number of steric clashes, that have fewer intermolecular interactions, and that have higher relative strain energy. While not ideal, I think PoseCheck gives the field a sobering view into what still needs fixing. Charlie ended his presentation optimistically, saying that better architectures, generative modeling techniques (e.g., guided diffusion), and of course better training data may help improve over the status quo.
I’d be remiss not to mention a similar work for docking-based methods (PoseBusters) as well as the GitHub link to PoseCheck.