

On Training Data for Bio AI Models
As we advance biological foundation models, which lessons from LLM data curation transfer, and which need rethinking?

Special thank you to Chris Gibson, Nathan Frey, Peyton Greenside, Ron Alfa, and several others for insightful conversations throughout the writing of this article.
Intro
One of the largest trends in AI this year has been the incredible spend by frontier labs on proprietary training data generated by external partners. While the labs’ spending metrics are rarely publicly disclosed, they can be estimated using numbers from the data labeling firms themselves. Back in June 2025, the largest players, Surge AI and Scale AI, were averaging ~$1B in revenue each, driven mainly by spend from the frontier labs.
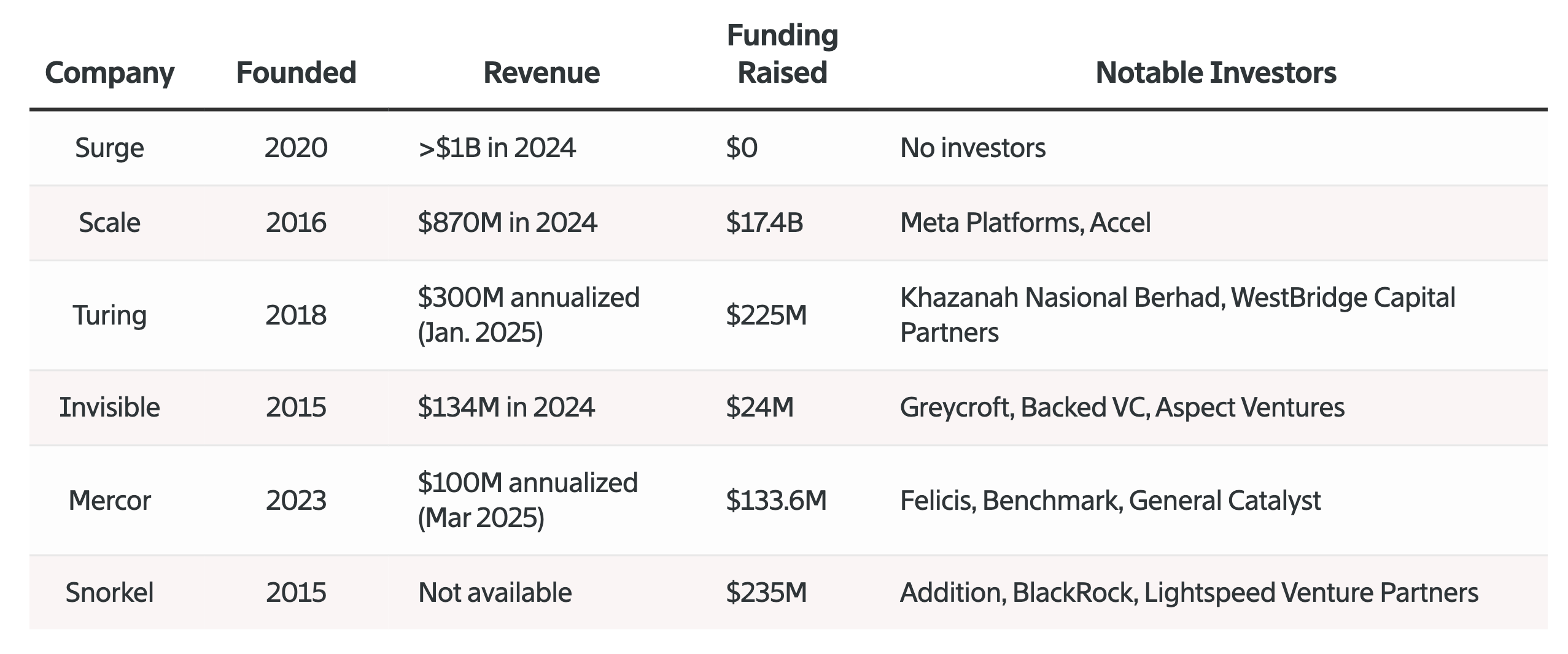
Extrapolating to the larger data market, it’s been estimated that top labs are shelling out $1-10B annually on proprietary dataset generation from several data providers. These numbers seem to only be increasing as bespoke datasets and reinforcement learning (RL) environments become cemented as core accelerants for improving model performance across disciplines.
If you simplify the axes of how large language models (LLMs) improve into (1) access to relevant training data, (2) advancements in model architecture, and (3) increased compute, this trend toward purchasing external datasets makes sense for a few reasons.
The combination of data collection efforts over the last several years (e.g., Common Crawl scraping the internet, data licensing from publishers) has accelerated the exhaustion of all existing human-generated text data.
Major advancements in model architectures are often commoditized over time (e.g., transformers) due to frequent talent transfer between labs and large investment into research, making architecture a less reliable moat for AI labs.
Compute constraints are increasingly governed by growing demand to serve models rather than by the compute needed to train them.
This data demand has manifested most widely across hot application domains like coding, finance, and computer use. The logic is rather simple, and the playbook seems to be working. Including more curated examples of Python code in your training data will likely make the LLM a better software engineer.
I am increasingly seeing these same trends discussed for the biological datasets used to train life science foundation models, with model developers making more licensing deals for access to task-relevant data. Including more high-quality antibody affinity measurements in your training data will likely make the generative protein model a better protein engineer.
There has also been a growing focus on improving the speed and rigor with which we collect these biological datasets to feed this demand. Autonomous life science robotics companies are now pitching themselves as data foundries to take advantage of this growing market in an effort to close the flywheel between data collection, model training, and evaluation.
These growing opportunities to collect, sell, buy, and train on biological data raise some important questions: what’s the right strategy for curating and training on datasets for biological foundation models, and what principles do and do not transfer from how we approached training LLMs with text data?
Despite the macro-level market parallels between frontier LLMs and biological foundation models, there are meaningful and nuanced differences that are worth discussing in depth.
From a data perspective, the historical playbook for LLM training — a predominant focus on data scale — will not transfer one-to-one to biological datasets. Instead, I argue that curating biological training datasets demands far more care in prioritizing data quality over quantity, and we should recognize this before we fall into the trap of scalemaxxing.
The LLM pretraining paradigm
In the early days of modern LLM pretraining, the predominant consensus was to improve model performance through scale. More data, more parameters, more compute. This manifested with the field spending much of its time enumerating pretraining scaling laws — the slope of how model performance improved as a function of data, model size, compute, etc. We wrote about this topic in depth in a previous Dimension Research article.

The general takeaway from this early period has likely been seared into the minds of many AI researchers: “More data gives you better results.” Scale could take priority over data quality because humans had spent centuries assembling a large bank of high-quality writing (e.g., excerpts from the internet, published literature) that LLMs inherited essentially for free. They were originally able to sidestep the canonical trade-off between data quality and quantity from the start because the inherited dataset contained enough of both to enable useful model performance.
Researchers could leverage scale while using high-level filtering approaches (e.g., deduplication, decontamination, early quality classifiers) to remove low-quality examples. Noise was further tolerated because text data is highly redundant, so random errors and contradictions average out at the scale we already had. Once models could generate their own high-quality synthetic data, the trade-off was reduced even more.
The field has started to see this playbook rebalance over the last couple of years. As we ran out of available data, the appreciation for high-quality datasets became another prominent lever to pull during pretraining, and researchers began to develop more aggressive data prioritization tools to enable this at scale.
Data Selection with Importance Resampling (DSIR) — From a large dataset, define a sub-selection of high-quality data. Then sample this “high-quality signature” from the broader distribution of the full dataset to surface all the other high-quality examples.
Domain Reweighting with Minimax Optimization (DoReMi) — Train small models with varying weights of different data types (e.g., 50% general knowledge, 25% coding, 15% science, 10% reasoning traces) to find the most performant distribution. Scale this distribution up to larger models once you find the optimal mix.
Model-Aware Data Selection with Data Influence Models (MATES) — Train a small “data influence model” that can dynamically select the most effective data for each stage of primary model pretraining to optimize the order of data the model sees.
Quality Classifiers (FineWeb, DCLM, Nemotron-CC) — Train separate classifier models to score data quality based on user-defined metrics and filter anything that doesn’t meet a quality threshold. Smaller but higher-quality datasets can be more performant than a larger dataset with mixed quality (e.g., a 7B DCLM model performed similarly to an 8B Llama 3 model with ~6x less training data).
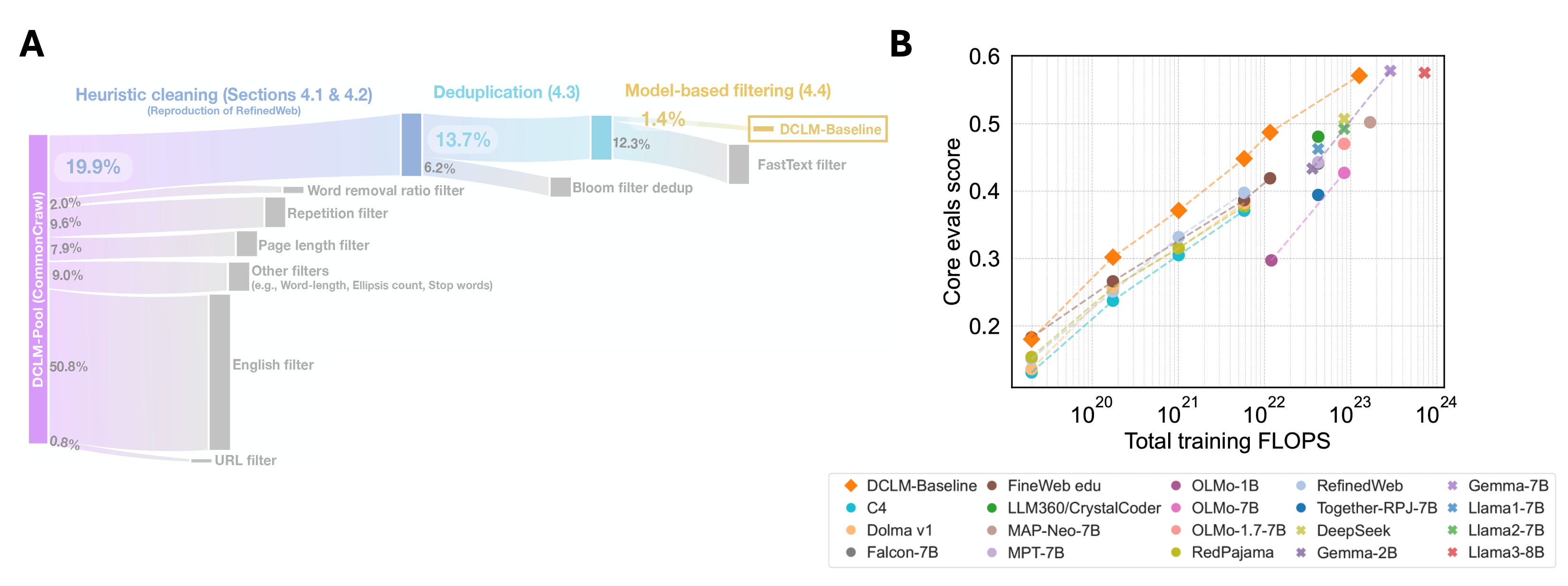
These approaches all showcase the newfound importance of data quality for LLMs, but most of them rely on having an existing large corpus of well-defined, high-quality data to filter from. Not a problem for LLMs a couple of years ago, but we are milking nearly everything we can from existing data.
The huge spending from frontier labs on application-specific data is further evidence that quality datasets across domains have become a major bottleneck.
Thankfully, we have a decent recipe for how to generally approach more data collection. For many areas of interest, it’s relatively easy to verify data quality and associated benchmarks (e.g., the code outputs the right answer, the mathematical analysis outputs the correct closed-form answer, the report on the history of tennis is factual and interpretable by a human). We may even be able to repurpose and build upon some of the above quality filter tools to prioritize which novel data to procure externally.
Unfortunately, this data framework for LLMs does not directly port to the world of biology.
Why biology breaks this playbook
Biological foundation models are rapidly progressing across a wide range of subdomains and applications. Biomolecular models are generating full-length, high-affinity monoclonal antibodies in silico, genomic foundation models are starting to predict variant effects and design realistic DNA sequences, and tissue-level world models are being developed to predict spatial protein expression and patient drug responses.
These achievements can be partially traced back to breakthroughs in deep learning and LLMs over the last several years. Many initial architectures and scaling intuitions inspired by text models were subsequently applied to public biological data.
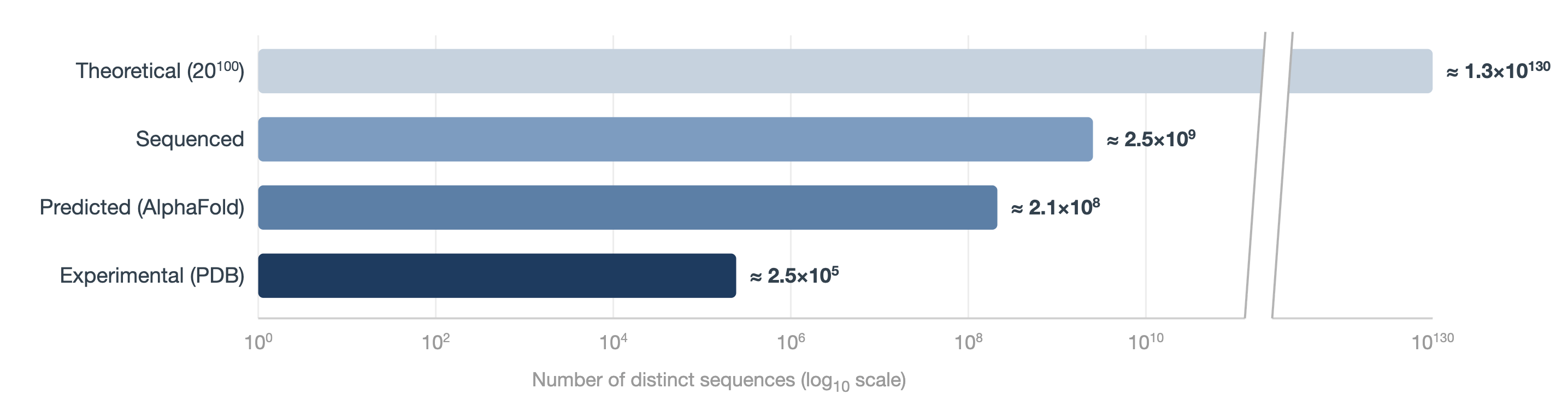
But this playbook only really works when the data is well-curated. The canonical example is AlphaFold, which showcased how pretraining on the Protein Data Bank (PDB) — a high-quality experimental dataset of ~200k protein structures curated over decades — could lead to incredible performance in structural prediction.
But the PDB is more of an exception than the norm. Unlike text data for training frontier LLMs, our publicly available biological data is much more sparse and suffers from several unique and meaningful limitations that make it difficult to use for model training. As a result, training performant bio foundation models often requires new data to be synthesized, and (bio) frontier labs are beginning to explore a data procurement strategy similar to the one we’re seeing with LLMs to access it.
In such a world, what training data is most useful to generate for a given application? What should be our philosophy on biological data collection? In the life sciences, where the trade-off between scale and quality is more apparent, how do we appropriately balance the two?
Answering these questions requires a more formal definition of what I mean by “high-quality” and an understanding of the limitations of current biological datasets compared to text training data. Here, I define quality as (1) context-rich, (2) clean (i.e., reduced noise and reproducible), (3) diverse, and (4) purpose-built to learn ground truth.
Context scarcity
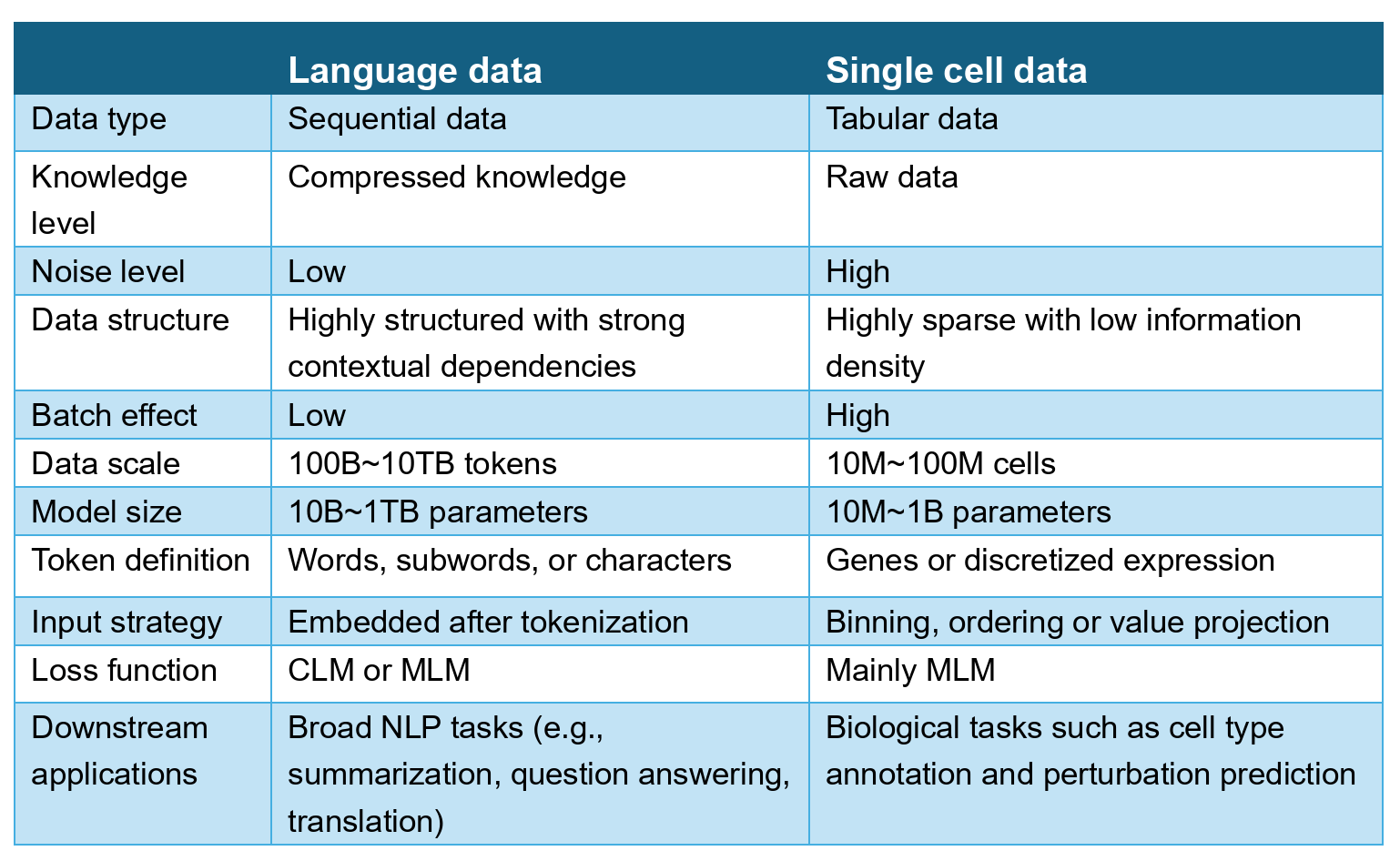
It’s hard to compete with the scale of more or less all recorded human history. Some of the largest LLMs with publicly reported metrics are trained on tens of trillions of tokens post-filtering. But my argument is not simply that we have more text data than biological data. We actually have a decent amount of biological data — tens of trillions of nucleotide bases, hundreds of millions of protein sequences, hundreds of millions of single-cell transcriptomic profiles, and hundreds of thousands of experimental protein structures.
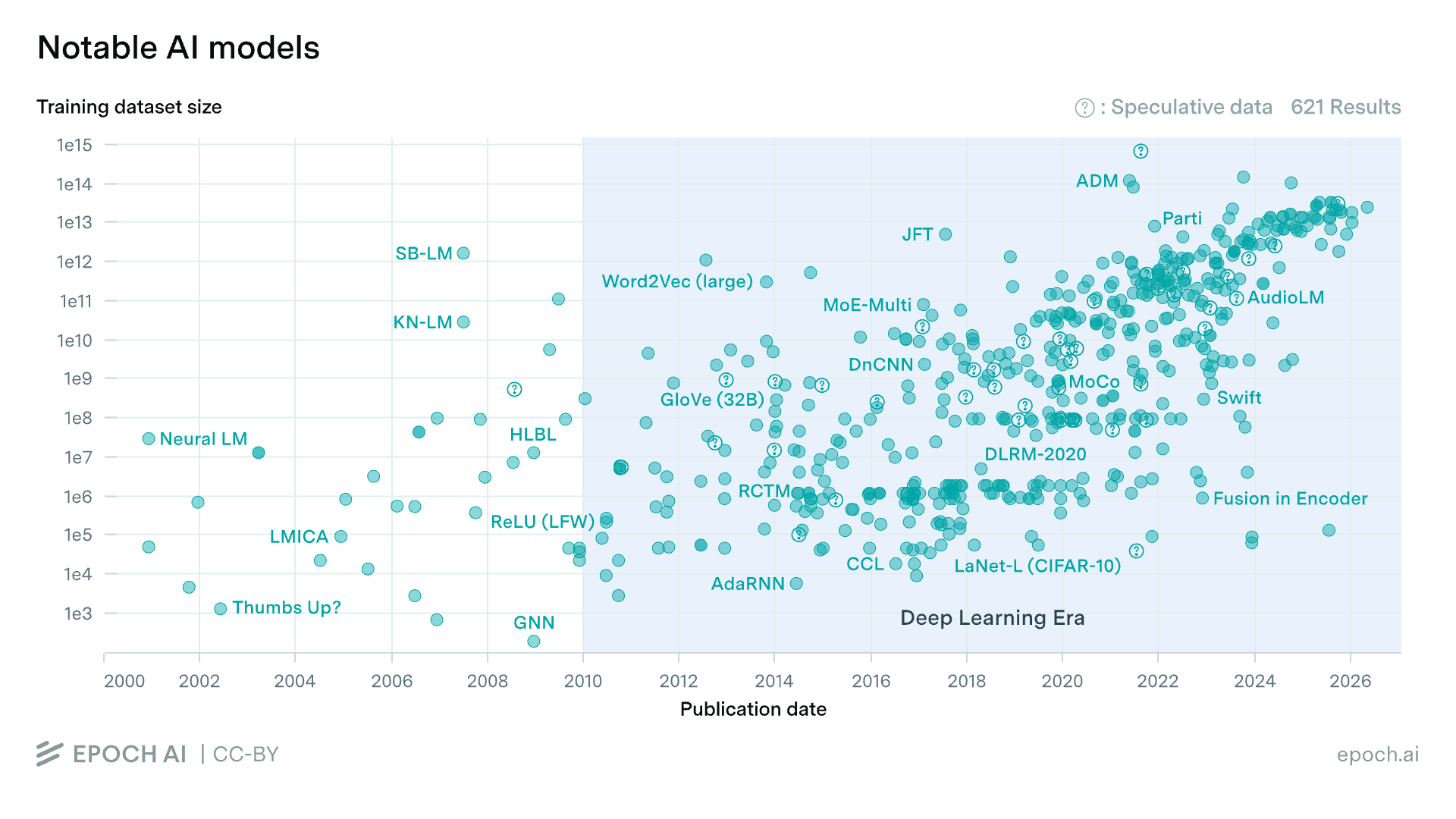
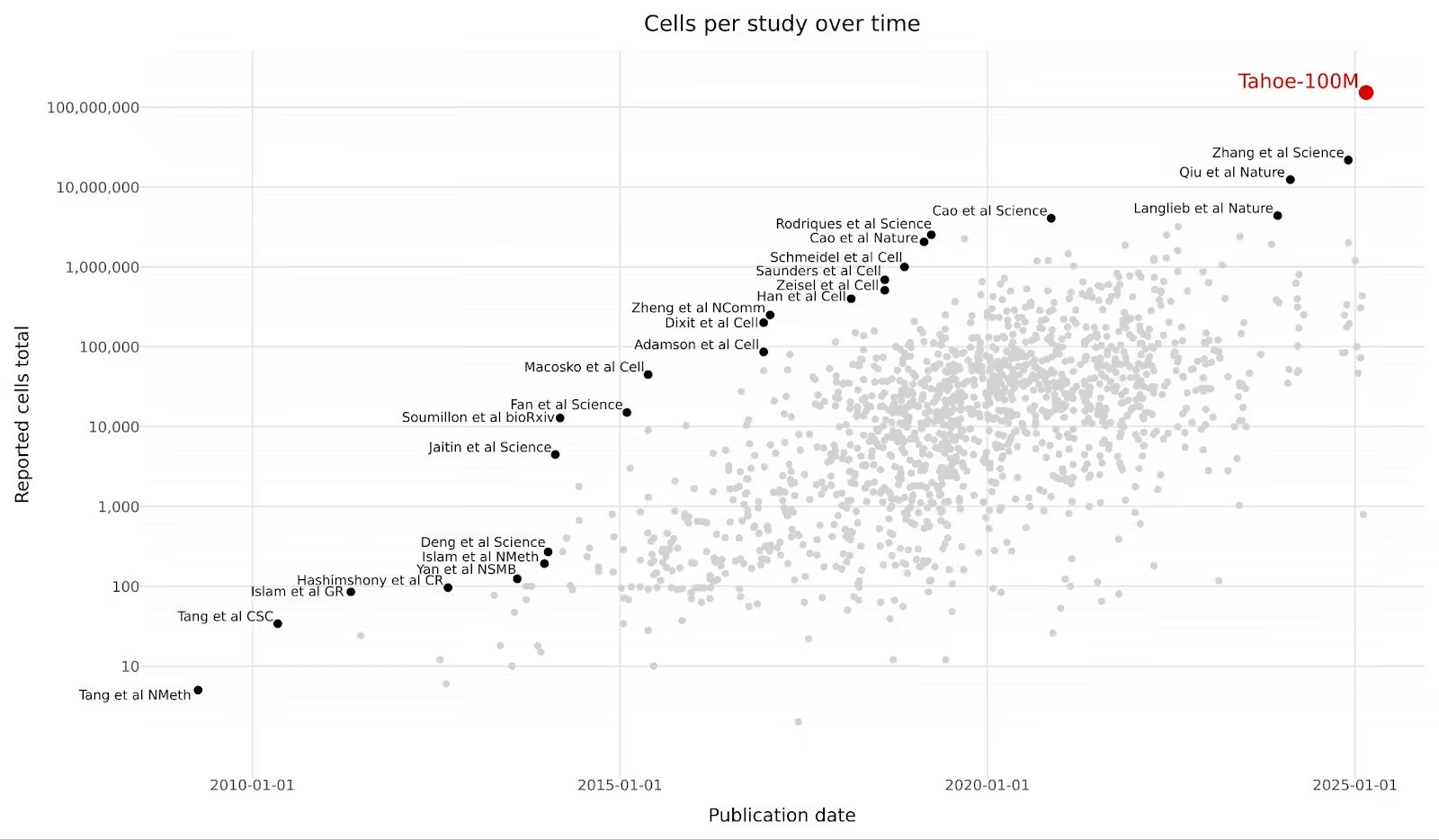
The more important difference between text and biological data is context scarcity.
With text data, the relevant context is already baked into language — sentence structures, document organization, back-and-forth conversations, logical topic explanations. All of these are human- and machine-readable without additional heavy annotation. This is particularly important for self-supervised pretraining via next-token prediction where researchers optimize for learning signal per token. For example, when you mask the next word in the sentence, the previous words and sentence structure natively provide the relevant context to predict what is missing.
Some biological data types, such as protein or DNA sequences, carry a similar type of inherent context to text data. For example, neighboring residues constrain which amino acids are likely present within a certain motif. We can even use a similar masked language modeling approach to learn these patterns in biological sequences and predict the missing amino acids.
One reason this works so well is that protein and DNA sequences contain native evolutionary context which we can extract through techniques like multiple sequence alignment (MSA) for extra predictive power. This works well when the property you are trying to predict is one that evolution was already selecting for. For example, a residue is often conserved because it’s relevant to protein function, so a model that learns a broad sequence distribution could theoretically predict whether a mutation results in loss of function without training on a specific labeled example.
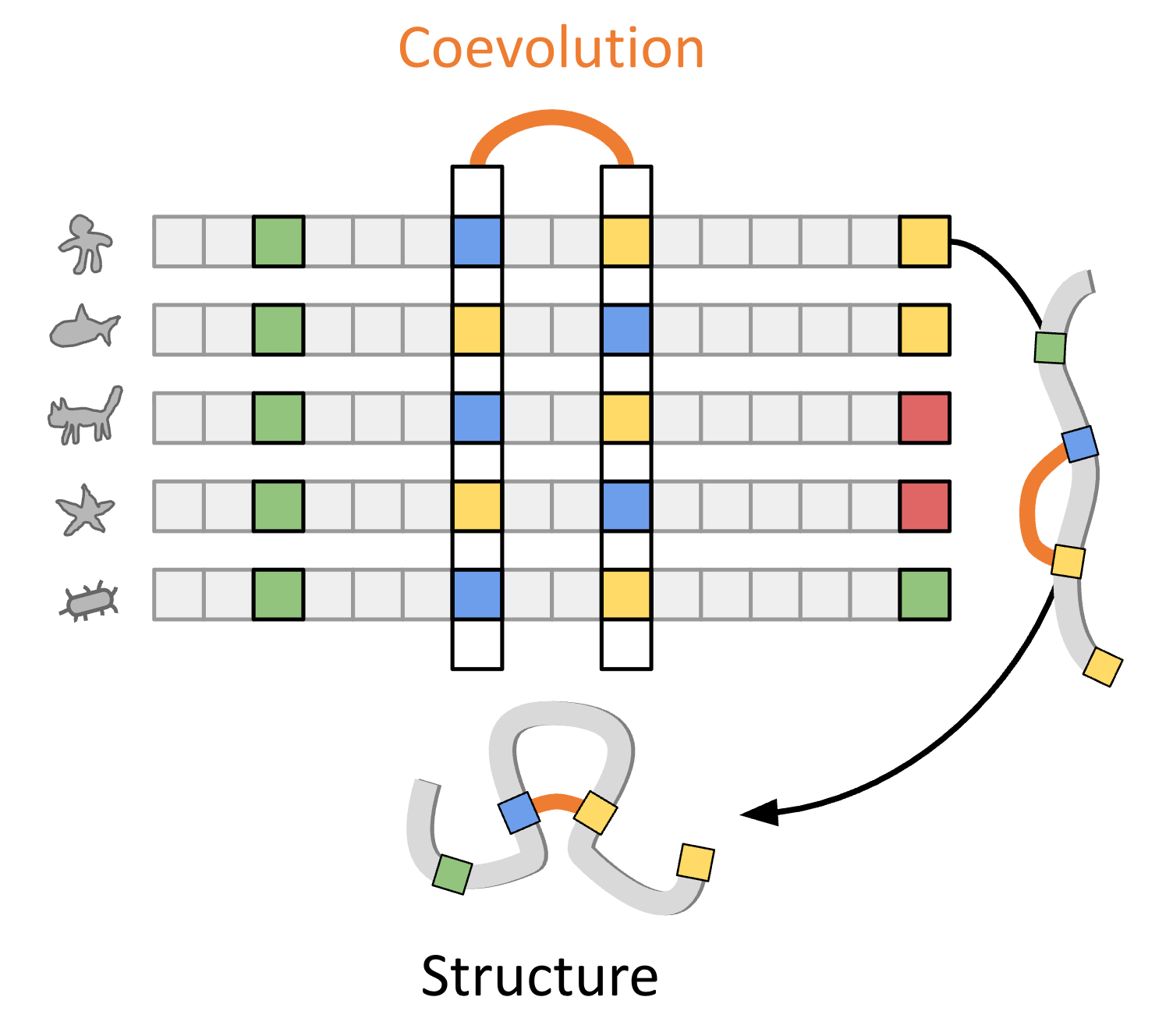
But evolutionary signal is limited, and we risk misinterpreting it if it’s the only context we rely on. For example, it doesn’t fully account for all the failed attempts in evolution (e.g., embryonic lethal mutations) or the lost or under-sampled organisms we don’t have data for. It also assumes conservation or co-variation implies importance, which isn’t guaranteed.
Maybe most importantly, evolutionary signal from native protein sequences alone doesn’t include other useful context specifically relevant to drug discovery like novel protein functions, binding affinities to synthetic targets, post-translational chemical modifications, selectivity across off-targets, or drug developability properties. When modeling more complex biology such as cell phenotypes, relevant context — cell genotypes, environmental conditions, and protocol-level details — becomes even more limited in current datasets.
All the above missing context exists in the physical world, and we have to painstakingly measure it experimentally and use those measurements to annotate the data. Moreover, generating high-quality, annotated biological data is often very expensive and requires a level of sophistication in assay design and execution that can be a significant roadblock to non-experts. The trade-off between cost, time, quality, and quantity is ever present for wet-lab scientists.
There is another related problem that underlies all of this. Biological context is limited to whatever we’ve already observed and/or discovered, which bakes in bias from the start. Thus, all biological datasets are incomplete, and humans may not have developed techniques to measure the missing context, let alone even understand what the relevant context is for annotation in the first place. New discoveries are constantly reshaping our view of biology and impacting which context we prioritize to measure — the realization of the diversity and impact of solid tumor microbiomes, the discovery that RNAs can be glycosylated to affect their localization and function, or the full end-to-end sequencing of the human genome that corrected errors in previous references and introduced ~200M new base pairs previously unaccounted for.
Staged pretraining is how the field has tried to get the best of both worlds and partially offset context scarcity in biology. The ESM family of models is a great example. Self-supervised pretraining on large unannotated protein sequence datasets produces strong base embeddings from inherent evolutionary signal, while sequential fine-tuning on smaller, well-annotated datasets layers in application-specific performance. While this approach is more efficient than training on labeled data alone, the downstream performance still heavily relies on the quality and context of the annotated data used for fine-tuning.
Noise
Even if we could curate a fully annotated, context-aware dataset, biological data is still inherently noisy and the true training signal can be difficult to cleanly extract.
You may be thinking that finding signal through the noise is actually something deep learning can do quite well. And you would be right in a lot of ways. Models trained on messy web data still learn fluent grammar and broad world knowledge because text is redundant and its noise is largely random and uncorrelated, resulting in a lot of it being averaged out at scale.
Biological noise is much more complex. It comes in a variety of flavors that are often impossible to deconvolve. This matters because some of this “noise” is actually moonlighting as useful signal that we want to reliably capture and learn from. For example, a core challenge in life science research is embracing biological “noise” (e.g., patient-to-patient heterogeneity or sub-clonal diversity in tumors) while reducing technical noise (e.g., shot noise in low-count measurements or proxy readouts with limited dynamic range) and systematic noise (e.g., user-to-user batch effects or protocol variations that stack across labs).
Separating these primary sources of variation allows us to more faithfully represent the diversity of biological samples, which is critical for real-world drug discovery tasks like predicting therapeutic response across patient populations. As a tangible example, I wrote a review article a few years back about this exact topic in the context of faithfully modeling human tumor samples using organoids.
A problem is that all of our measurements in biology introduce both technical and systematic noise, some more loosely understood than others. Oftentimes these noise signatures are strong enough to mask the actual biological signal, making it highly susceptible to accidental bias. Fortunately, technical noise can more easily be averaged out at scale, but this is not true for systematic noise. Models can end up associating certain cell phenotypes with which lab generated the data, which instrument was used, or even whether the sample was located at the edge of a well plate versus the center, instead of learning the underlying biology that actually distinguishes them.
For example, collecting enough data for training bio foundation models often means curating related datasets across labs into a central, public repository (e.g., CELLxGENE for cell expression data, ChEMBL for molecule bioactivity). Due to systematic noise, there are considerable batch effects which get folded into the model’s representation of biology, and more data can make this even more prominent if we’re not careful.
One study even found they could train a classifier model to predict the chemist who synthesized a molecule using just ChEMBL structure data, since labs often pursue characteristic scaffolds that leave recognizable signatures in the molecules they produce. The authors then showed that chemist identity was enough to predict bioactivity nearly as well as a model with the structural information, suggesting that a model could learn to “cheat” its way to predicting molecular activity without fully learning the core structure-activity relationship.
By contrast, the techniques used to generate and collect text data are near-perfect. We can hire world experts to write reports on new subject areas and use web scraping tools to extract existing digitized text. We even have ubiquitous tools like autocorrect and spellcheck that de-noise our datasets in real time before they ever reach a training run. Moreover, the text data filtering tools I mentioned above rely on existing large datasets that already contain high-quality data, which is not always true in biology.
Diversity
Current AI models are truly exceptional when prompted with tasks that are in-distribution for their training dataset; however, performance significantly degrades when the model is asked to generalize outside of this distribution. Thus, a major effort across domains is to expand training data diversity to put as many relevant examples in distribution as possible.
But data diversity doesn’t just refer to one thing. It can be (crudely) represented by two major sub-genres: stylistic and semantic. For text data, stylistic diversity describes when the same underlying content is expressed in variable ways — the same fact presented casually, formally, across languages, in a verbose or concise way. On the other hand, semantic diversity describes when the underlying content expands the core information represented by the dataset — different facts across different topics. The former ensures the model is steerable and doesn’t get locked into a single style for writing a passage or a single method for solving a problem, while the latter ensures that the model can perform well across a broader range of subdomains.
These same genres also map loosely to biological data. Stylistic diversity is analogous to measuring a similar biological output in different ways — the same binding affinity measured in two labs or the same protein structure measured by crystallography and cryo-EM. Semantic diversity expands the data distribution by measuring completely new biology or annotations — new protein sequences or functional attributes.
Similar to the argument about context scarcity above, biological diversity is far more limited in our current datasets than it is in text, and if not properly accounted for, these two sub-genres pose their own unique risks to advancing biological models.
For example, stylistic diversity in biology is a double-edged sword. If done properly, increasing the diversity of data collected across protocols or techniques may provide beneficial signal that results in an underlying model that is more robust to variation. But if we’re not careful, stylistic diversity becomes uniquely entangled with biological noise and risks the introduction of batch effects that actually detract from model generalization.
Semantic diversity has also been lacking in current datasets for a couple of reasons. Many of our largest biological datasets are mined from biological samples that occur naturally: protein sequences in UniProt, protein structures in the PDB, genomes in GenBank. For practical reasons, these datasets still only represent a small slice of what naturally exists, which in itself is a very small slice of all possible biological diversity. Evolution is limited by survivorship bias and is not sampling all possible designs. This becomes a constraint in areas like generative protein design because many drugs we want to develop may look nothing like what has occurred naturally.
In addition to an evolutionary bias, the diversity of our current biological datasets is heavily biased by scientific interest and what’s fundable. Data is often curated in an attempt to answer very specific biological hypotheses, rather than to future-proof for training ML models.
Synthetic data generation is one area being explored to rectify limited data diversity. It’s the idea that when a model’s outputs reach high quality, they become useful for generating their own training data. A recent study from Meta found that supplementing real training data with synthetic text can match or even improve on the performance of LLMs trained only on real data. But it’s a slippery slope. Synthetic data quality needs to be high, and over-indexing on synthetic datasets can lead to model collapse, where performance is significantly degraded.
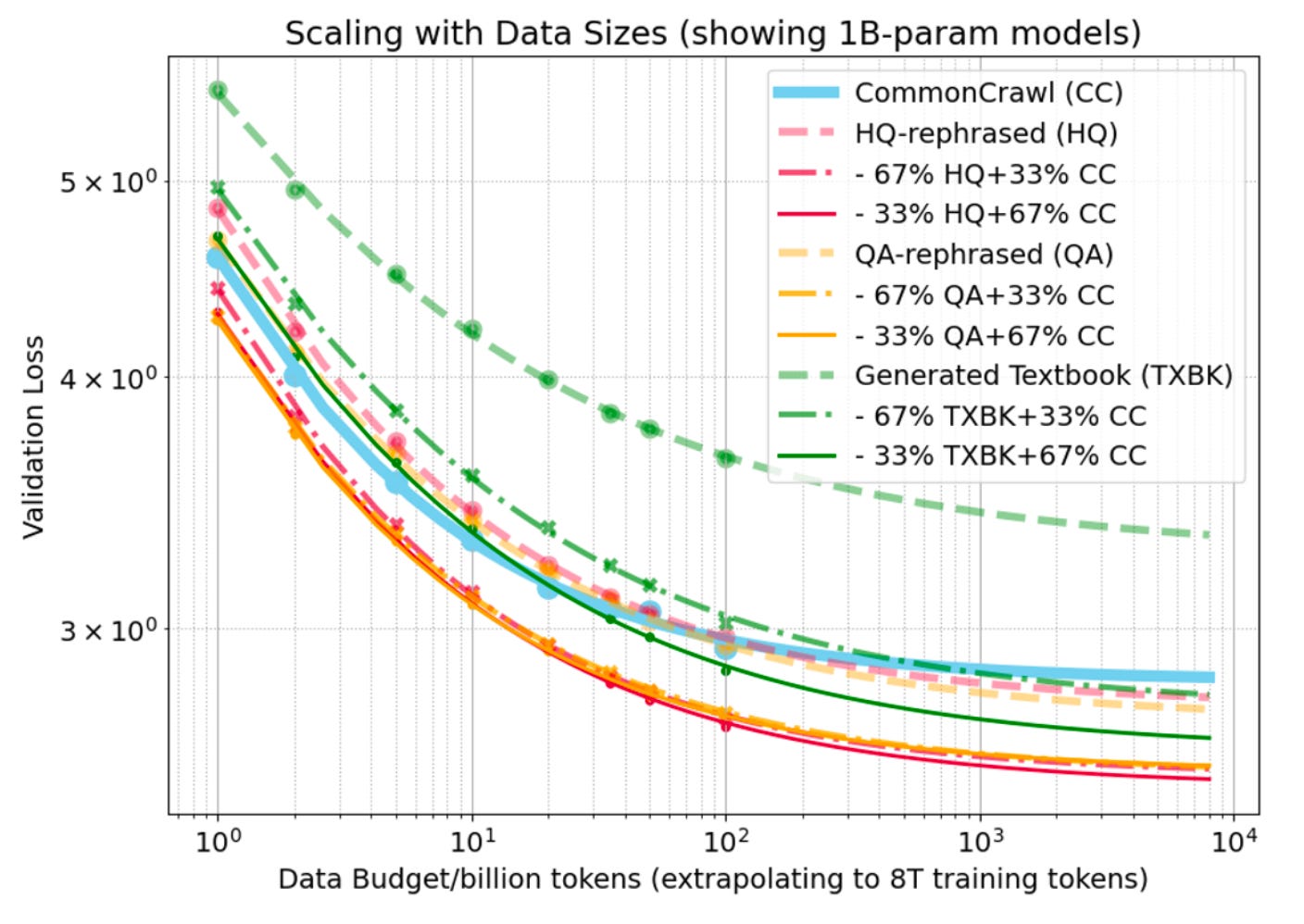
As you may expect, the current benefits of synthetic text and biological data start to diverge.
For tasks like writing, synthetically generated text can provide valuable training signal and is inherently “correct” as long as it generally makes sense to the reader. It’s often very easy for humans (or even LLMs themselves) to know when synthetic text quality is good or bad just from reading it.
For biology, where verification is inherently difficult, synthetic biological data is only “correct” and meaningful if it matches experimental outcomes from the lab. It’s easy to take a generative model and design millions of plausible-looking protein structures, but their real function or binding affinity remains unknown until they are measured in the lab. Any synthetic labels generated will simply inherit the bias or batch effects from the model that generated them, and without consistent verification, you risk training on artifacts of flawed models versus the real biological signal.
We have seen some early success with synthetic data generation in very specific biological applications. For example, AlphaFold 2 was trained with self-distilled synthetic data generated on sequences not present within the PDB, leading to increased performance across this expanded sequence space. This strategy generally worked because structure predictions from AlphaFold 2 reached a quality bar to be useful for training, and there is a clear way to verify these structure predictions in the wet lab if needed.
Ground truth
This last quality metric builds directly from the previous three. Even with meaningful context, clean signals, and diverse data, biology is still immensely complex, and it’s often extremely difficult to verify ground truth.
Within any domain, reliable output verification is a central mechanism for completing the data feedback loop and improving the model via reinforcement learning. As I mentioned at the beginning, fields like coding often have verification baked in, which has been a central reason why coding agents have grown exponentially in performance over the last year.
But in biology, ground truth has to be measured experimentally, which is much more expensive, complicated, and time-intensive than executing code. These measurements are also often proxies for the actual signal we are trying to record (e.g., photons for measuring expression of fluorescently tagged proteins), and these signals are usually only linear in small windows of dynamic range. We also don’t have a true understanding of our measurement’s positive predictive value toward human efficacy for drug discovery applications.
These limitations have prompted the field to think creatively about what scaling biological data should look like. A recent paper on black-box data scaling makes several convincing points for a future framework. In the paper, the authors argue that we can’t rely on human readability of datasets or heuristics to reach the scale we need to train performant biological foundation models. Instead, data sources need to be optimized for “machine consumption rather than human intuition.”
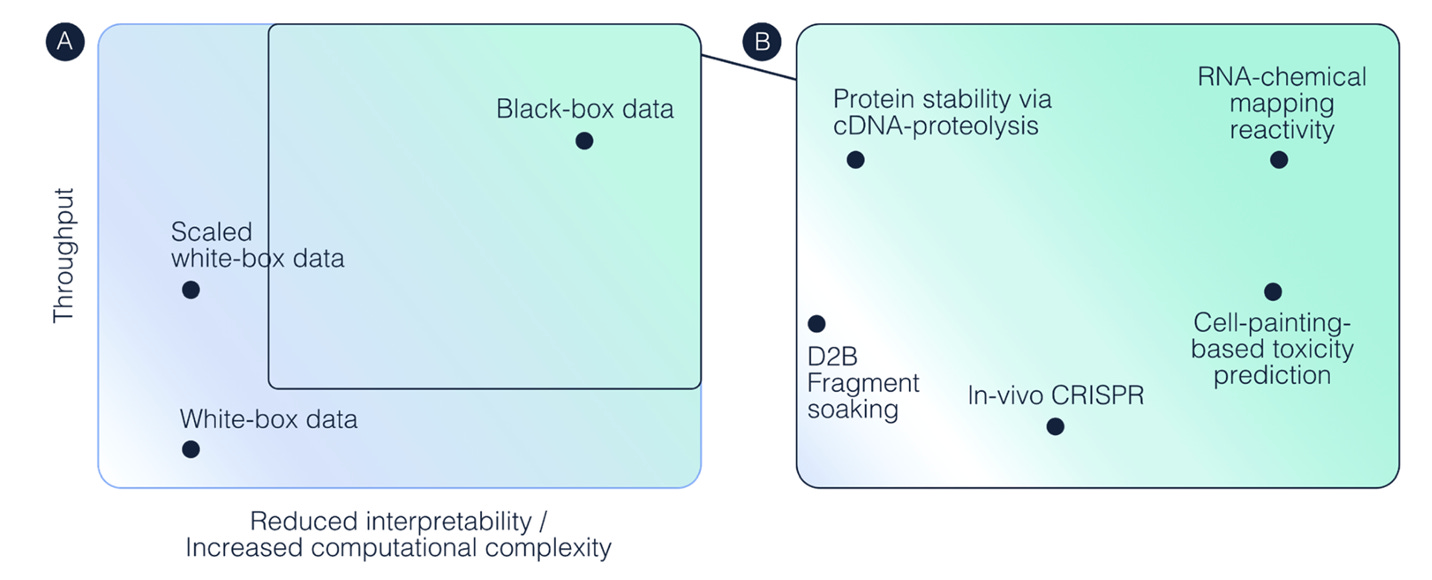
Despite much of my article’s framing, I think we will eventually start to embrace this philosophy more, but I argue we need to first have a balanced emphasis on building a playbook for maximizing data quality and biological understanding as well.
A representative example is the rise of “virtual cell” models trained on transcriptomic data to predict cellular response to perturbations. The training data is often drawn from a narrow set of immortalized cancer cell lines, skews toward specific tissue types, captures expression data at only a single snapshot in time, and combines single-cell measurements across protocols with different depths and noise profiles. Despite these datasets including up to ~100M cells, several independent benchmarks have shown these models struggle to outperform much simpler approaches on the downstream tasks most relevant to drug discovery.
The goal is not to crudely reduce a whole subfield to these practices. There is a range of data used across virtual cell models, and a lot of important infrastructure and exploration has been achieved pushing these models forward. But this example is a useful reminder that scaling data volume can outpace scaling data quality, and that biases and gaps in context often get baked into the model in ways the model itself cannot see. Therefore, scaling too quickly without the proper verification, biological insight, or quality controls could result in us scaling black-box models in the “wrong direction” without even knowing. And given how slow the verification loop is in biology, it may take a lot time before we realize it.
What this means for model developers and data generators
The four vignettes above argue for a modified playbook for collecting and training on biological data compared to text. Scaling without close attention to quality won’t get us there. We need a tight loop between methods for generating high-quality, task-relevant training data, evals that can accurately validate that data, and models tested against real-world tasks across the life sciences — ideally combined in a flywheel fast enough for each round of validation to inform the next round of data design.
This is much easier said than done, and there are many great teams executing toward these goals on both the model development and data generation sides — and many on both. Here is where I see the most opportunity for both camps.
For model developers
Just as we have seen for frontier LLMs, bio model developers are becoming increasingly data-hungry for novel training datasets to improve benchmark performance. As we enter this market dynamic, three increasingly important questions should be top of mind for developers: (1) how to assess data quality from providers, (2) how to verify its effect on real-world benchmarks, and (3) when to scale given limited resources.
In this future, data procurement starts to resemble a scientific peer-review process more so than a typical exchange of capital for assets. Which data is being purchased matters just as much as the metadata on how that data was collected and annotated. Protocol-level details, consistency metrics, batch structures, and replicate handling all paint a more complete story of the data package relevant to assessing data quality and training more reliable, context-aware models.
Given the complexity of biology and quality being measured on high-dimensional axes, the current quality classifiers, batch correctors, and filters used for text data will likely not port over to biological models directly. Developing biology-specific toolsets should be a major priority for model developers — or even third parties that sit between model developers and data generators — to scale this process.
We are already seeing examples of this in production. scVI, a tool which was originally developed for batch correction of scRNA-seq data, is now also being used as a quality filter to detect when dataset signal is being driven by user batch effects over the fundamental biology.
Model developers also need to validate performance through evals, but building robust evals for biological models is its own challenge due to the points discussed above. Additionally, held-out test sets often share the same batch effects as the training data, which means benchmark performance often reflects learned artifacts rather than learned biology.
Today’s most common evals are designed to match the complexity of the model. An antibody design model is likely to have a suite of benchmarks on affinity, selectivity, functionality, aggregation, and more. But as model outputs become more complex, validation outputs also become more bespoke, and the timescales and costs for this data grow increasingly large. At this point, more model developers may increasingly develop internal wet labs or make deeper partnerships with data generators that decrease time to validation in a more controlled, verticalized flywheel.
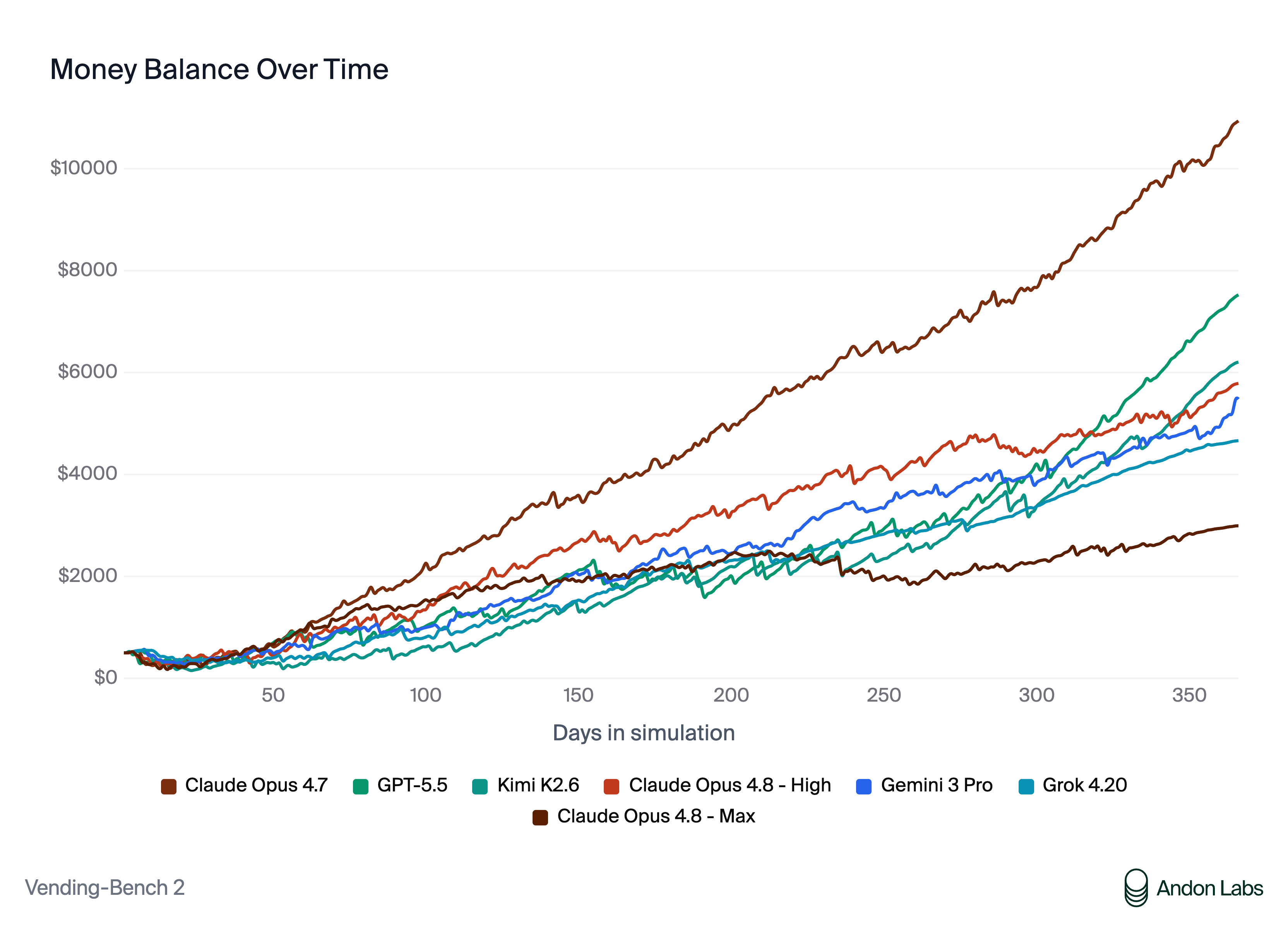
We need to start pushing validation as close to the real-world applications as possible. Andon Labs is a fascinating example of this for LLMs. They put agents in charge of a real vending machine business at Anthropic and a physical shop in San Francisco where success is graded on actual economic outcomes that are much harder to game than an artificial benchmark.
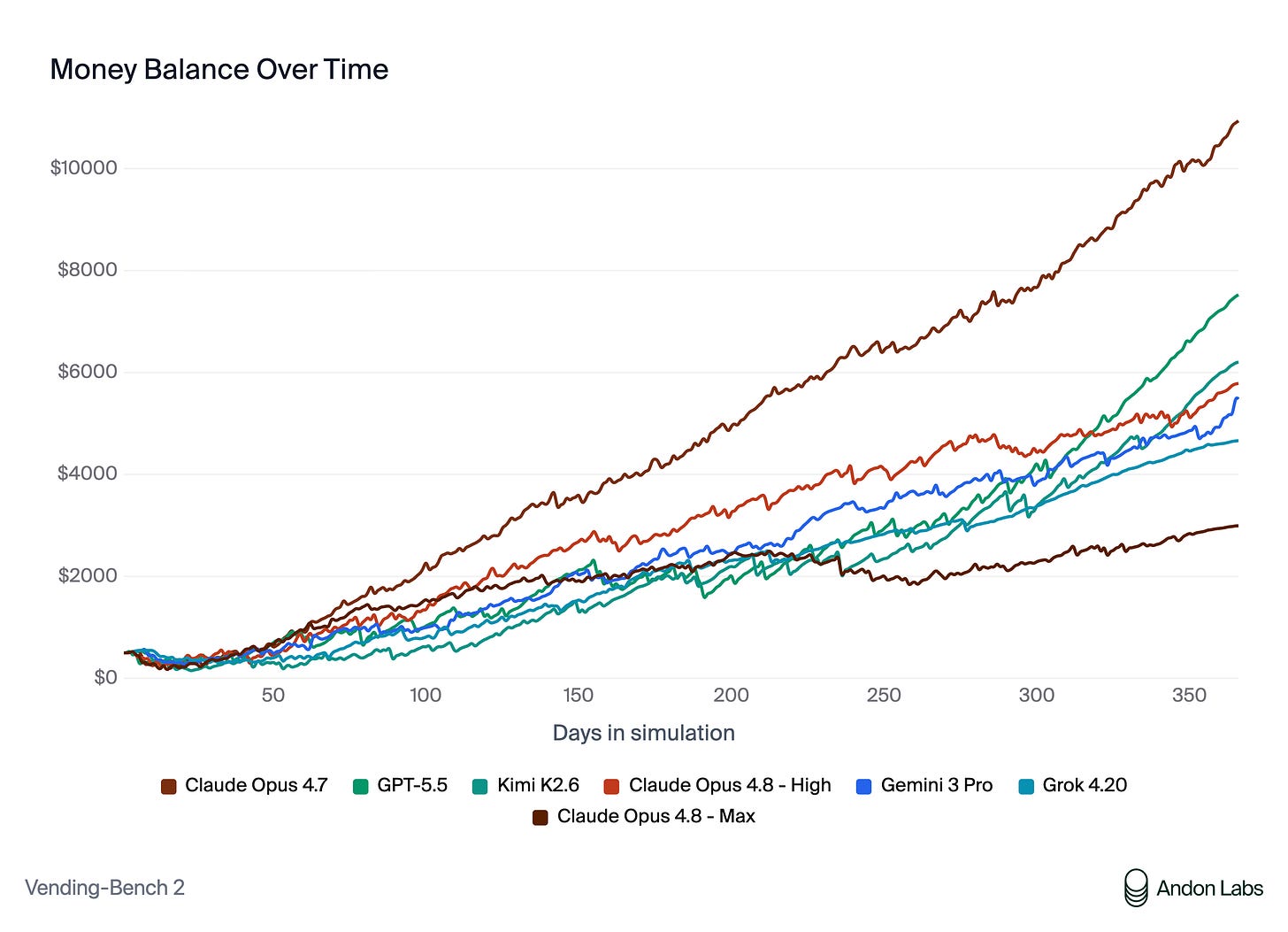
Results from the Vending-Bench 2 eval from Andon Labs. AI models were given a starting budget of $500 to autonomously manage a virtual vending machine business. The graph shows cash balance over time for each model. (Source) The equivalent of this for bio models is likely the drug discovery process itself, with model performance ultimately graded on whether its outputs survive preclinical and clinical readouts. The challenge is primarily cycle times, as drug discovery takes many years compared to daily or weekly economic feedback for a vending machine or retail shop. Nonetheless, the intention still stands, and the goalposts of clinical success as the end-all, be-all metric have not changed.
It’s likely impossible to design an assay or data collection strategy that addresses all four points raised above. We are constrained by finite budgets, short timelines, and limited technologies. In such an environment, developers need to understand when the right moment is to advance data collection protocols, and when to scale.
There is no single right answer here, but scaling should only start to happen when developers are confident the data is actually representative for the downstream task. For example, spatial transcriptomics provides more context than the scRNA-seq transcript counts alone, but spatial data is considerably more expensive and time-consuming to collect than scRNA-seq. If your primary use case relies on understanding localization between cell types within a tissue, this extra context is a worthwhile trade-off to prioritize.
Several other important considerations include: (1) an understanding of how much learning signal is present per sample, (2) what sources of noise are dominating data collection and how to best mitigate them, (3) the time and cost required to acquire the data, (4) how quickly you can verify the data once it’s collected, and (5) the cost of being incorrect and how reversible the scaling investment is if so.
To make this more complex, different data types will undoubtedly require different scales of data. Training on 100 cancer patient cases annotated with clinical outcomes may be better for some applications than training on 10,000 patient cases annotated with surrogate biomarkers that only loosely correlate with the true endpoint.
There is a real chicken-and-egg dynamic here. We may not learn the right biology without scale, but scaling too fast risks confidently indexing to incorrect or incomplete biology. The hard truth is that you don’t know how good your data actually is until you train a model and start to find out. This tension argues for careful consideration around both quality and scale from the start, rather than treating quality and biological insight as something we can simply filter on after first scaling.
For data generators
Biological foundation models are becoming increasingly complex and will require considerably more data than what is available publicly or what can be generated by model developers internally. Even Lilly — the most valuable pharma company on Earth with decades worth of data collection — has formed a data-sharing platform to meet the high volume, diversity, and quality of data needed to develop AI models for drug discovery.
Data generators come in many flavors: (1) a biotech translating a high-throughput screening platform that generates unique insights into biology, (2) an automation/robotics company working to scale the actual workflows of valuable assays, (3) a CRO that has refined data collection for customers over years, or (4) even a new company category dedicated to optimizing data collection specifically for model developers.
My argument is that those players who increasingly focus on high-quality data and who perfect the data handoff to model developers will be the most successful. Their value proposition to the community is multifold, but I will focus on three in particular.
The first and simplest value proposition today is having a platform for generating unique biological data. This could be attributed to a novel screening platform or to pushing a validated platform into a new therapeutic niche. Arguably the most overlooked piece here is the exhaustive annotation of datasets and the infrastructure needed to implement it, from data-specific attributes down to which user is running the experiment. The platform, the data, and the annotations contribute to the moat.
Without heavy investment in annotation and the required infrastructure, this value proposition is fleeting. Assays — even complex ones — are often commoditized over time, and players can be leapfrogged by new technologies or updates to the techniques by others. These players can also win early on price and time-to-data, but there is a high risk of racing to the bottom with their competitors.
As model developers become increasingly savvy at diligencing biological data opportunities, a reputation for generating high-quality data adds to this moat. Data generators with documented, traceable protocols, machine-readable metadata, proper batch controls, sufficient replicate measurements, and consistent annotation quality become trusted sources in the field, which translates directly into pricing power. Despite how interesting or unique the underlying biological data may be, poor data quality and lack of organization compound, and every quality improvement provides higher leverage.
The third and most mature value proposition begins to change the shape of the relationship between the data generator and model developer. Today’s default mode for CROs is predominantly reactive. They get customer inbound to run an experiment, they collect and ship the data, and they collect their fee. The economics are often driven by which vendor can meet the minimum quality specifications for the cheapest price in the shortest amount of time. Another race to the bottom.
Data generators have unique insights into the limitations of current models that the developers don’t even know themselves. This insight starts to unlock new value when data generators (1) move upstream to proactively scope which datasets are likely to improve a model’s evals, and (2) generate their own evals to demonstrate the worth of their data on specific tasks.
As high-quality data becomes a stronger moat to model developers, this relationship starts to look more like a dedicated commercial partnership than a data-vendor relationship, which likely commands even greater and lasting economics. I also imagine developers will prefer exclusive licenses to datasets, making this tighter relationship even more important to cultivate from both ends.
Parting thoughts
Training data has always been at the center of progress in AI, no matter the subdomain. This is especially true for biological foundation models that come with their own unique considerations.
It’s clear there is not going to be a one-size-fits-all approach to how we scale and train biological foundation models, and that should be welcomed by the community. Due to the sheer complexity of biology, each subdomain will require its own model architectures, tokenization strategies, and considerations for how exactly to balance data quality and scale.
None of what I have laid out here is an argument against scale, so much as it is an argument for high-quality, diverse data. We can’t make meaningful progress in digitizing biology with only one or the other. In a world of only scaling our current understanding of biology, we miss critical context that we may not have even discovered or learned to measure yet. In a world too focused on perfecting datasets, we lose out on our ability to develop meaningful models to advance drug development today. The end goal is still translating meaningful assets to patients as soon as possible.
I’ve often heard the phrase “quantity is its own form of quality,” and I generally agree with the sentiment, but my point is that this statement is only true in biology when the data is high enough quality in the first place. As the field increasingly starts to build more performant biological models aimed at some of the toughest problems on the planet, the data bottleneck will be solved by not solely focusing on “how much data can we collect?” but “what data should we be collecting in the first place?”
One of the best recent pieces I’ve read on landscape for developing biological foundations models. It’s comprehensive and generally follows my intuition as well when it comes to modeling real world complexity. Thank you
An incredible depth. Thank you for putting time into it. For platform builders, it's worth going through Bauer's checklist. Makes sense and is detailed.