

NeurIPS 2024 Open Notes
Our team visited Vancouver to attend NeurIPS 2024. These are our initial notes from many of the AI x Biology workshops.


Forward
Conferences like NeurIPS are quite useful for staying on top of fast-changing fields such as computational biology. Unfortunately, not everyone who might like to can attend. Similar to our recent notes from MoML, we wanted to share our scribblings from the weekend workshops at NeurIPS 2024. Below, you’ll find notes from selected talks during four workshops. We hope you enjoy this resource!
Please feel free to write us with any comments, questions, or corrections!
Contents
Advancements in Medical Foundation Models
Machine Learning in Structural Biology
GenAI for Health: Potential, Trust, and Policy Compliance
Advancements in Medical Foundation Models
Workshop Summary
“There have been notable advancements in large foundation models (FMs), which exhibit generalizable language understanding, visual recognition, and audio comprehension capabilities. These advancements highlight the potential of personalized AI assistants in efficiently assisting with daily tasks, ultimately enhancing human life. Despite the great success in general domains, FMs struggle in specific domains requiring strict professional qualifications, such as healthcare, which has high sensitivity and security risk. In light of the growing healthcare demands, this workshop aims to explore the potential of Medical Foundation Models (MFMs) in smart medical assistance, thereby improving patient outcomes and streamlining clinical workflows. Considering the primary clinical needs, we emphasize the explainability, robustness, and security of the large-scale multimodal medical assistant, pushing forward its reliability and trustworthiness. By bringing together expertise in diverse fields, we hope to bridge the gap between industry and academia regarding precision medicine, highlighting clinical requirements, inherent concerns, and AI solutions. Through this cooperative endeavor, we aim to unlock the potential of MFMs, striving for groundbreaking advancements in healthcare.” —Workshop Website
Speaker Summaries
Current Advancements and Roadblocks for Foundational Models in Pathology and Ophthalmology; Pearse Keane (University College London, Moorfields Eye Hospital)
Foundation models for applications in pathology and ophthalmology was a pervasive theme throughout the workshop, with three excellent speakers rounding out the morning session with their insights into the current advancements and challenges in this rapidly growing space.
The first talk was from Pearse Keane, a Clinician at Moorfields Eye Hospital and Professor at University College London, who is leading a multidisciplinary group of ophthalmology researchers at the intersection of health and AI.
The ophthalmology field is quickly becoming overwhelmed by increasing patient populations seeking treatment, and has surpassed orthopedics as the #1 busiest specialty in terms of clinical appointments in the NHS in the UK.
Medical foundation models trained on vast amounts of ophthalmic images could help alleviate some of this demand by helping to more efficiently and effectively diagnose patients, especially for diseases like age-related macular degeneration (AMD), which is one of the top causes of blindness in older populations.
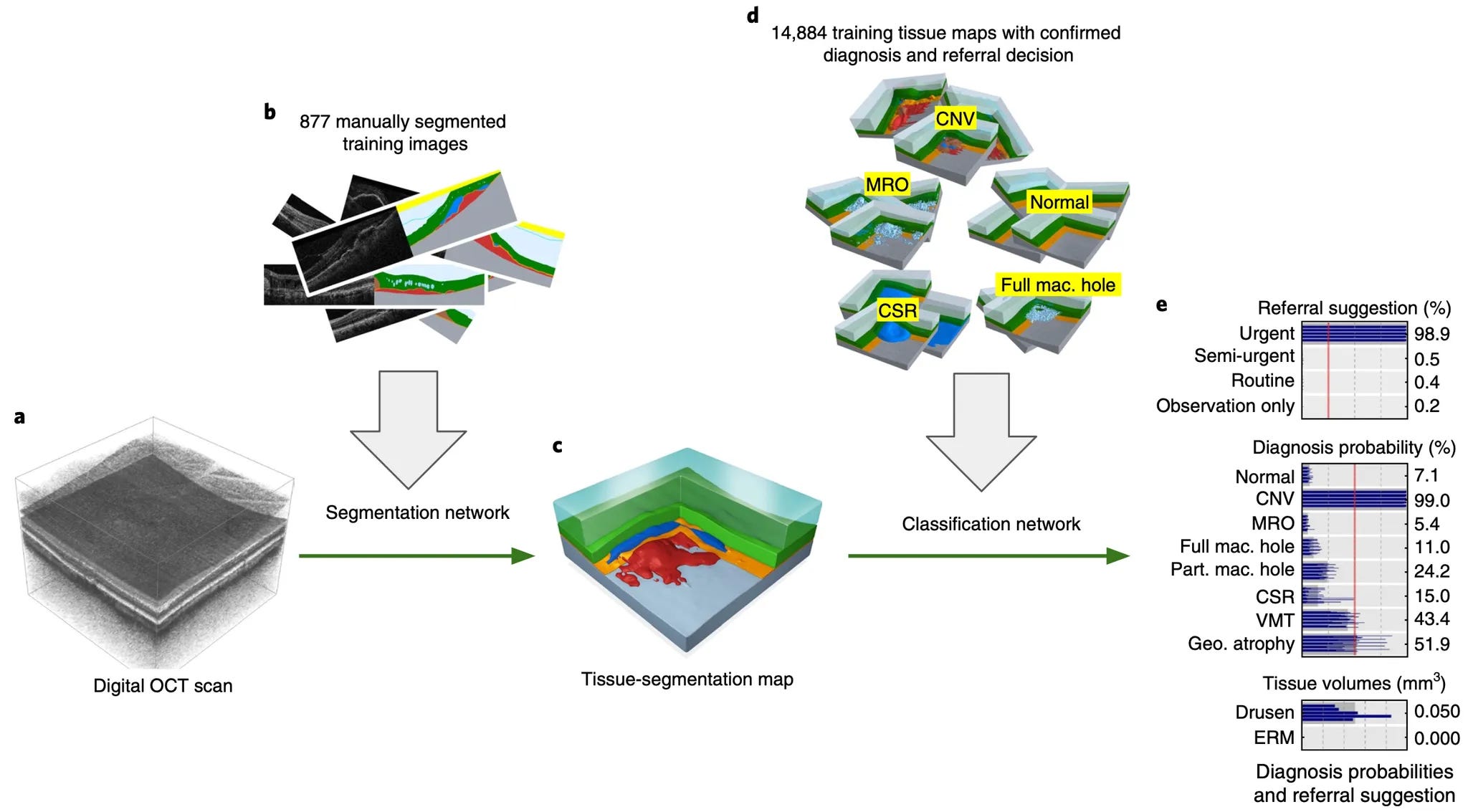
To this end, Prof. Keane highlighted several efforts he and his group have been developing, including a collaboration between Moorfields Hospital and Google DeepMind. Together, their model was trained on 1.2M anonymized retinal optical coherence tomography (OCT) scans to identify some of the most common causes of blindness. More details on the research can be found in their Nature Medicine paper.

An AI-based framework to diagnose patient with select eye diseases and suggest referral urgency for followup. A digital OCT scan is segmented and classified using two distinct networks that result in a probability of having a specific diagnosis. (Source)
Faisal Mahmood (Harvard Medical School)
The second talk was from Faisal Mahmood, a Professor at Harvard Medical School, who is developing generative models for applications in pathology.
Whole slide images (WSI) of histological samples contain a vast amount of biological information given the high resolution of digitally-scanned slides.
Prof. Mahmood and his group have developed several models trained on datasets of WSIs to predict patient diagnosis, prognosis, treatment response, survival, biomarker expression, and many other characteristics. More recent work has focused on building foundation models supplemented by other biological data including special stains, genomics, and transcriptomics to augment their ability to predict the above properties.
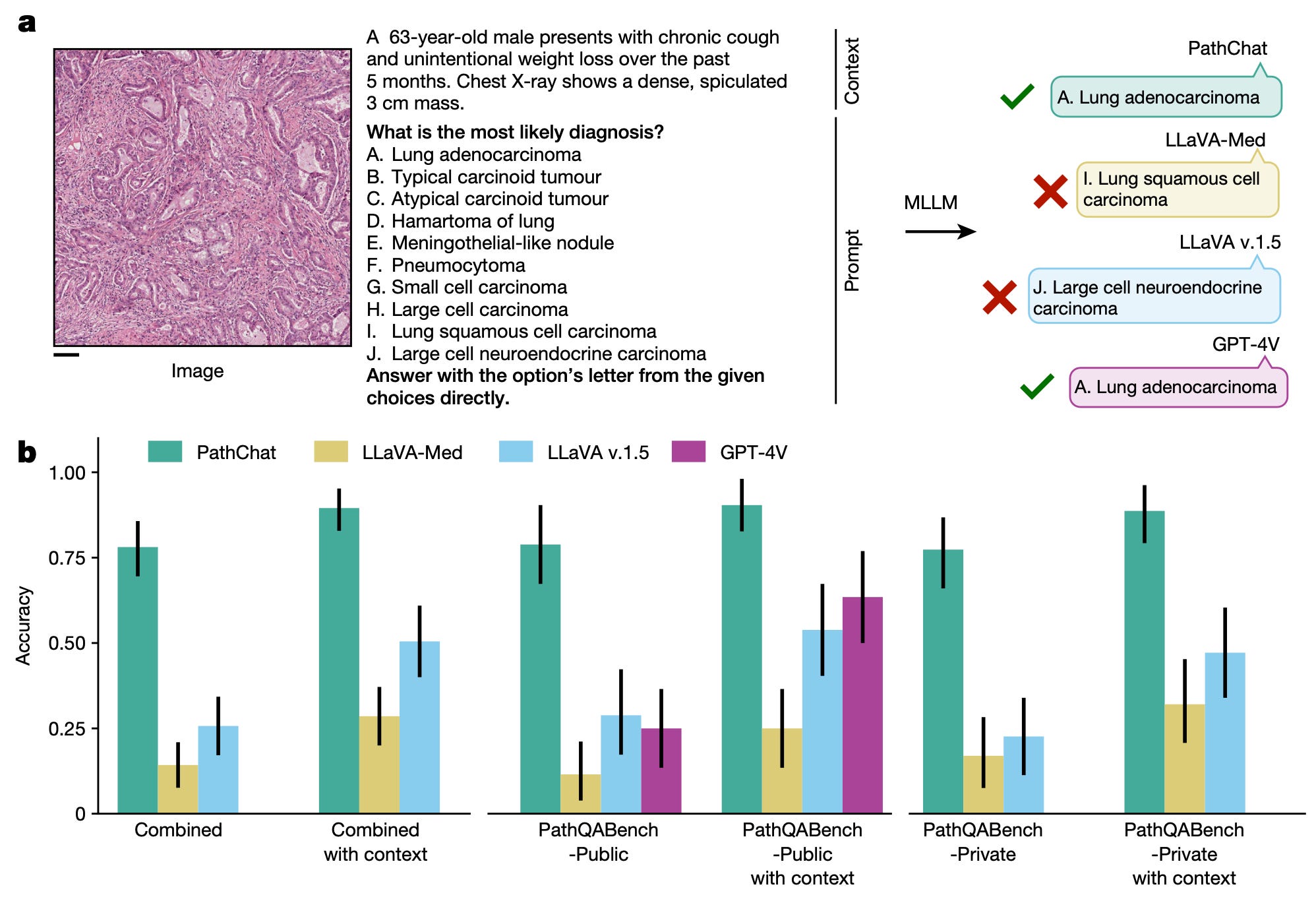
In addition to this work, he presented on a strategy to develop a generative AI copilot for applications in pathology analysis. The model, called PathChat, is a natural language chatbot-style interface between the user and their pathology data which enables the ability to ask questions like: “What tissue type is shown in this image?”, “Based on this image, what is the likely diagnosis for this patient?”, and “Can you synthesize a pathology report for this sample?” Check out their recent Nature paper to learn more about PathChat!

A benchmarking evaluation comparing PathChat to several other multimodal LLMs in their ability to diagnose a patient based on a pathology slide and minimal textual information in multiple choice format. On average, PathChat outperformed the other models on 105 questions (the Combined data) with an average accuracy of ~75%. (Source)
Sheng Wang (University of Washington)
The third talk was from Sheng Wang, a Professor at the University of Washington, focusing on building generative multimodal foundation models for the biomedical sciences, including ophthalmology and pathology.
Similar to PathChat, the team developed their own foundation model called GigaPath, which was trained on ~171k pathology slides from more than 30k patients covering 31 tissue types. They showcased the ability for GigaPath to use chain-of-thought to better generate patient treatment plans by first predicting disease subtype, then predicting associated biomarkers, and finally a recommended therapeutic plan. More details on GigaPath can be found in their recent Nature paper!
In the final portion of his talk, Prof. Wang introduced their work on multimodal foundation models incorporating data from many distinct imaging techniques (e.g. ultrasound, pathology, X-ray, OCT, etc.). To achieve this, they implemented BiomedParse, a separate foundation model also developed by their group, to project data from 9 different imaging modalities onto text space. Effectively, this can cluster images representing similar phenotypes or tissue types together in text space making it easier for the model to learn from multiple image modalities at once.

BiomedParse can accurately segment medical images from several different imaging types with Dice scores > 0.9 (i.e. a measure of how well the BiomedParse binary mask overlaps with the ground truth mask, where 1 is complete overlap). (Source)
Overall, there were several major overlapping themes throughout these three talks that set the scene for future of biomedical foundation models.
Low adoption rates amongst experts in these fields can limit the development and usefulness of these foundation models. How do we best introduce new technologies into clinical settings that augment the work currently being performed by clinicians?
Foundation models are beginning to ingest a substantial amount of data across hospitals, institutions, and other clinical care sites. However, there is considerable bias introduced when combining datasets from disparate sources. These biases can be attributed to different personnel, instruments used for data collection, varying protocols, among many others. How do we sufficiently address these biases? The room’s consensus seemed like this problem may need to be addressed through both changes in model development and the way we collect and store our clinical data.
Foundational models require well-curated datasets. Traditionally, clinical data has not been sufficiently organized in a systematic manner to enable rapid integration into novel foundational models. A major focus moving forward will be working with care providers and institutions to standardize data storage with a particular focus on patient privacy.

AI + Science: Applications to Medicine; Anima Anandkumar (California Institute of Technology)
Prof. Anandkumar opened her talk with a call to the community to consider more than just LLMs when building foundation models: “Foundation models tend to be synonymous with language models to many of you, but it’s not only natural language. I may hint a little bit about it, but most of my talk will not be about language models.”
This quote set the stage for her presentation where Prof. Anankumar focused on the compelling idea that when building biological foundation models, we need to consider how to implement a truly multi-scale approach with a diverse repertoire of data.
However, building these types of foundation models can be very costly because traditional methods have been based purely on numerical simulations of biology. These simulations can be very computationally expensive even for our most advanced supercomputers.
Prof. Anandkumar and her group have been developing AI surrogates to replace these numerical simulations at many biological length scales: from quantum modeling of individual molecules, to simulating viruses, to building the first AI-based high-resolution weather model.

Overland wind speed prediction by FourCastNet AI weather model (top) compared to the true wind speed data (bottom) for North America. The FourCastNet model relies on a neural operator architecture to reliably incorporate and forecast weather given datasets with variable resolution. (Source) To do this, their group introduced the use of neural operators. Unlike neural networks that learn to map finite inputs to outputs, neural operators learn how to map infinite dimensional function spaces. This means they are well situated for these types of multi-scale modeling tasks because they can simultaneously capture the complexity of different input processes at different resolutions.
For example, global weather patterns might look grainy if you zoom into the street-level view, but they become more useful when zooming far out. But in the same model, you may also want to include high-resolution mappings of fog data on the level of a single city.
In contrast to neural networks which typically have input and output data at a fixed resolution, neural operators are a useful tool to incorporate data at multiple resolutions.
Prof. Anandkumar went on to give several examples of how her lab has leveraged neural operators in their work, including for MRI and photo-acoustic image reconstruction from a wide variety of subsampling patterns.
For the final portion of the talk, Prof. Anandkumar highlighted a recent set of projects from her group detailing how the AI technologies they developed can directly impact clinical applications.
Of note, one example included training surgical robots on multimodal feedback data collected during surgeries (e.g. video and audio captured from a camera mounted to the robot) with the ultimate vision to use these AI robots to help train the surgeons themselves.
Another application that she highlighted was the potential medical insight that could be gleaned from training AI models on wearables data (e.g. Apple Watch tracking your steps, sleep, heart rate). They are particularly interested in developing energy-efficient analog neural nets for electrocardiogram (ECG) arrhythmia classification.

Levels of Clinical Evaluation for LLMs; Karan Singhal (OpenAI)
Carefully-executed evaluations of medical large language models (LLMs) are critical to their successful deployment to solve real-world problems.
(Medical) LLMs are quickly approaching a regime of “increasing capability overhang” meaning these models are much more capable than what we are currently testing and using them for.
Prior to deploying these models in real-world settings, we need a system of evaluations that enable researchers to build, test, and tune LLMs in a medical-specific context.
In his talk, Karan systematically outlined a framework of 4 levels of clinical evaluation for LLMs and gave examples of published work from each: (Level 1) Automated Assessments, (Level 2) Human Ratings of Open Generations, (Level 3) Offline Human Ratings for Real Tasks, and (Level 4) Real World Studies.
Level 1: Automated Assessments:
This first level of assessment often leverages the use of multiple choice questions (e.g. MedQA) in a similar style to how a medical student would be examined. This is often a measure of the model’s underlying knowledge and reasoning abilities, including questions around clinical diagnoses.
These evaluations tend to be relatively easy to execute and have fast iteration cycles; however, they lack the ability to test for real-world behaviors, often do not have a wide dynamic range to stress-test models, and suffer from contamination with model training data.
Interestingly, consumer LLMs (e.g. ChatGPT) tend to perform quite well on these tasks despite not being fine-tuned with specific medical knowledge for clinical applications.
Level 2: Human Ratings of Open Generations:
The second level of assessment involves the model producing a longer, open-form response to a wide variety of questions, which is subsequently rated by a human expert. Typical tasks can include medical diagnoses from a list of symptoms and patient messaging services. These outputs are often compared directly to a physician, and to avoid reviewer-to-reviewer bias, the reviewers select the best answer from options derived from several LLMs.
These evaluations enable targeted measurement of model knowledge and behavior and tend to be more relevant to real-world situations than Level 1; however, biases in human ratings are difficult to control for, including biases towards output length and formatting, resulting in the need for carefully-crafted rubrics for grading.
Level 3: Offline Human Ratings for Real Tasks:
The third level of assessment is structurally very similar to Level 2, but with a focus on real-world tasks that clinicians would perform regularly. This could include using a vision language model for producing radiology reports or electronic consultations between doctors.
These evaluations are advantageous because they can provide really useful performance metrics on real-world tasks without the need for a costly real-world study; however, they still share all the same limitations of a Level 2 study (i.e. human bias and standardized rubric design).
Level 4: Real-World Studies:
The fourth and final level of assessment brings LLMs to a fully real-world context with real clinicians and patients directly or indirectly interacting with the model.
There have been relatively few Level 4 studies run to date, but Karan highlighted one recent paper that tested ChatGPT’s ability to assist with clinical documentation compared to human dictation.

Data summary of a blinded, randomized, controlled study benchmarking ChatGPT against other common techniques used by clinicians to document a patient’s history of present illness. Given its real-world deployment, this work is considered a Level 4 benchmarking study. (Source) These evaluations are considered the gold standard for their clinical relevance and strong comparators to current workflows; however, they often suffer from low sample sizes, costly and slow evaluation cycles, and continued bias amongst human raters.
We are just at the beginning of productionizing these models in real-world settings. Many opportunities exist for current researchers to both build novel evaluations and test their LLMs using this proposed framework.

Machine Learning in Structural Biology
Workshop Summary
“Structural biology, the study of the 3D structure or shape of proteins and other biomolecules, has been transformed by breakthroughs from machine learning algorithms. Machine learning models are now routinely used by experimentalists to predict structures to aid in hypothesis generation and experimental design, accelerate the experimental process of structure determination (e.g. computer vision algorithms for cryo-electron microscopy), and have become a new industry standard for bioengineering new protein therapeutics (e.g. large language models for protein design). Despite all of this progress, there are still many active and open challenges for the field, such as modeling protein dynamics, predicting the structure of other classes of biomolecules such as RNA, learning and generalizing the underlying physics driving protein folding, and relating the structure of isolated proteins to the in vivo and contextual nature of their underlying function. These challenges are diverse and interdisciplinary, motivating new kinds of machine learning methods and requiring the development and maturation of standard benchmarks and datasets.
Machine Learning in Structural Biology (MLSB), seeks to bring together field experts, practitioners, and students from across academia, industry research groups, and pharmaceutical companies to focus on these new challenges and opportunities. This year, MLSB aims to bridge the theoretical and practical by addressing the outstanding computational and experimental problems at the forefront of our field. The intersection of artificial intelligence and structural biology promises to unlock new scientific discoveries and develop powerful design tools.” —Workshop Website
Overview
This Sunday at NeurIPS, the Machine Learning in Structural Biology workshop showcased just how far the field has come since AlphaFold 2's release four years ago. The packed room and poster-lined walls reflected a field that's earned a Nobel Prize and continues to evolve rapidly. Here are some key developments that caught my attention, though this is by no means comprehensive or chronological.
As is natural for a discussion about machine learning, the talks naturally split between advances in models and datasets.
AlphaFold 3
Starting off with models, Josh Abramson, a research engineer at DeepMind and the lead author of the AlphaFold3 (AF3) paper, provided an inside look into its development. We’ve previously written an in-depth guide to AF3, so I will gloss over the details, but the major advancement from AF2 to AF3 was expanding the representation to encapsulate the whole PDB — including DNA, ligands, and ions.
This required two major changes: standardizing the input and generalizing the structure model. AF3 keeps the per-residue or per-nucleotide token representation for proteins and DNA but switches to a per-atom basis for other molecules and performs additional processing upfront to produce a generalized input representation. To generalize the model, they replaced AF2’s structure model that predicted spatial frames and torsions to a generative all-atom diffusion model conditioned on the preprocessed inputs.
Interestingly, AF3 removed equivariance (molecule invariance to rotation/translation) with seemingly no loss in performance. This was a fascinating contrast to a talk a day earlier by Johann Brehmer from Qualcomm research who suggested that equivariance improves data efficiency, though they suggest that data augmentation can close this gap if training for a long time. Yet in the limit of infinite data, equivariant models are still more compute efficient than non-equivariant models.
One notable challenge for AF3 emerged with unresolved PDB coordinates. Since AF3’s diffusion model is only trained on ground truth data, for unresolved or disordered regions it would predict unphysical, bunched up structures. To address this, the team generated a “disorder” cross-distillation set using AF2 predictions on these unresolved coordinates in the PDB, allowing AF3 to nearly match AF2’s accuracy. This exemplifies the extensive dataset curation required for AF3’s development. Josh concluded his talk discussing AF3’s limitations including missing protein dynamics or physics, hallucination, and chirality mismatch.
Boltz-1
Jeremy Wohlwend offered a complementary perspective on AF3 through the lens of training Boltz-1, an open-source, accessible AF3 replication. His talk revealed how the subtle implementation details often omitted from papers may explain why replicated models sometimes exhibit unexpected behaviors — for instance, Boltz-1 sometimes incorrectly superimposes very large identical chains, a behavior that may not exist in AF3.
While most of the model architecture and training sets remained the same, Boltz-1 does introduce new optimizations like a new confidence model that mirrors AF3’s trunk to improve confidence predictions. Additionally, they also constructed multiple sequence alignments (MSAs) with MMSeq2 and Collabfold, a significantly faster MSA calculation from Milot Mirdita and the Martin Steinegger Lab at SNU (who spoke earlier), allowing full MSA computation of the entire PDB on a single node within weeks. As an aside, the field owes a lot to researchers like Milot and Martin for highly optimizing these fundamental computational functions and tools. We are excited to see how others continue to innovate from on top of these models such as Chai-1’s recent introduction of model restraints to further improve accuracy.
Conditional Modeling
While structure prediction was once the primary challenge, the field has pushed towards conditional generation or designing proteins with specific functions with releases like RFDiffusion or AlphaProteo.
Noelia Ferruz, a group leader at the CRG, kicked off the day presenting fascinating work on generating enzymes with a specific function using reinforcement learning. In collaboration with BaseCamp Research, her approach began with training a protein language model (ZymCTRL) on protein sequences labeled with catalytic functions. The zero-shot model could generate some initial functional and diverse carbonic anhydrases (35% success rate) and further fine-tuning on BaseCamp’s metagenomic datasets greatly increased success rates and resulted in active enzymes at extreme conditions.
Yet, these models produced enzymes that largely mirrored the training set properties, which is not conducive to generating out-of-distribution properties. Her group then took a really interesting agentic view of protein design (a broader trend this year at NeurIPS) to address this limitation. This agentic approach iteratively rewards good sequences while penalizing poor ones and maintaining reasonable deviation from the original model. With this framework, her group guided the model towards a diverse set of alpha carbonic anhydrases rather than only the beta variants. They also applied this framework to the Adapytv Bio Protein Design competition (Dimension is a co-host). With limited optimization, they generated multiple successful binders for EGFR, with two surpassing EGF’s affinity.

Another great talk from Amy Lu touched upon the debate between sequence and structure models (see our recent MoML coverage). She addressed this with PLAID, a method to directly sample from a joint structure-sequence distribution to generate all-atom proteins. Her key insight came from examining ESMFold's inputs - the zeroed-out pairwise input into the structure head suggested that ESMFold’s sequence embeddings alone could represent both structural and sequence distributions. Intuitively, this makes sense as ESMFold maps sequence to structure, so its intermediary latent space should contain representations of both. Since sequence databases are much larger and more richly annotated, PLAID is able to generate proteins conditioned on functional and taxonomic inputs. While initial attempts to train a diffusion model on these embeddings failed, developing and adding the CHEAP encoder to compress the embeddings (inspired by high-resolution image synthesis) enabled generation of diverse, high-quality proteins across varying lengths. Amy showed that PLAID was able to generate a variety of diverse all-atom proteins conditioned on both function and taxonomy at many sequence lengths. These generated proteins impressively retained conserved active site motifs or membrane hydrophobicity patterns consistent with their conditioned input function.

Datasets
Switching gears to datasets, earlier in the conference, Ilya Sutskever declared that data is the fossil fuel of AI, and that we’ve exhausted this precious resource. Yet these talks at MLSB suggest that biological research continues to yield novel datasets and clever generation techniques.
Improving Databases
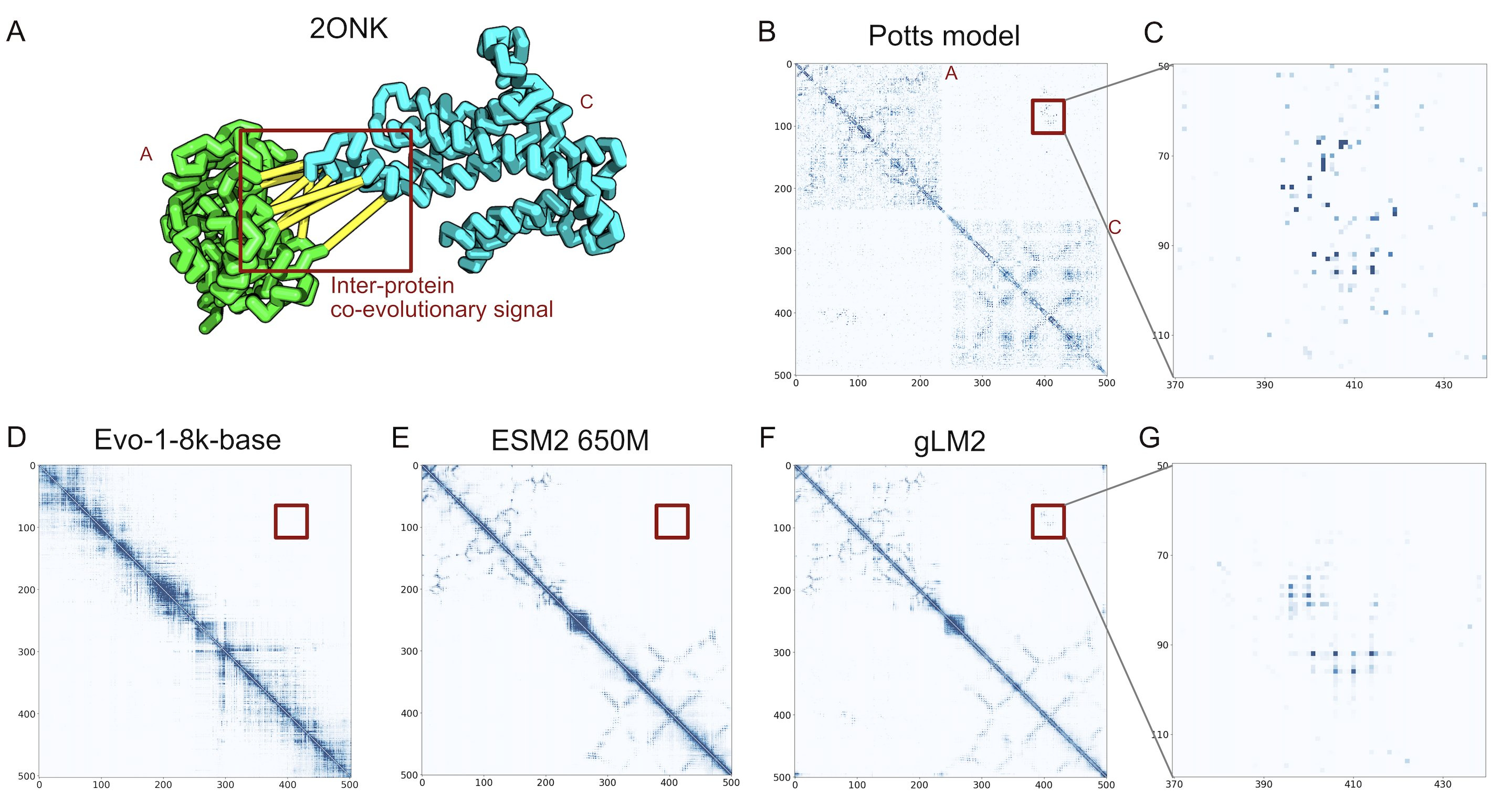
In this vein, Andre Cornman, the CTO of Tatta Bio, spoke about aggregating new sources of data through their Open MetaGenomic (OMG) corpus. He suggested we could think of these sequences measured from our environment as nature's raw dataset - like how Common Crawl scrapes the entire web, while UniProt is more like Wikipedia's curated entries. These metagenomic sequences are captured with their full genomic context intact through contigs. The resulting database contains 3.3B proteins, offering 3x more diversity than UniProt. To demonstrate the value of the additional data, they train a genomic language model, gLM2, on this expanded dataset. gLM2 outperforms ESM2 on the Diverse Genomic Embedding Benchmark and they showed that it captures co-evolutionary signal of a protein-protein interface without an MSA that both ESM2 and Evo miss.
Sai Advaith Maddipatla and his co-authors explored how to better constrain structure predictions with experimental data. Focusing on X-Ray crystallography structures, they developed a method to handle regions where proteins show distinct alternative conformations ('altlocs'). While these altlocs have been cataloged by crystallographers, current models only model one of these altlocs. Their team developed a method to calculate the expected electron density from predicted structures and use that to guide a pre-trained diffusion model (Chroma) by minimizing the difference between calculated and experimentally observed densities.
With this guidance, the model successfully generates ensembles of conformations that match experimental densities - accurately predicting multiple distinct conformations in flexible regions while maintaining precise predictions for stable ones. This work provides a way for protein structure prediction models to account for some of the inherent dynamics and flexibility present in proteins. One of the interesting future follow-ups for the team involves refining the PDB, potentially creating a more accurate training resource for future models.

Generating More Data
Gabe Rocklin, a professor at Northwestern, presented on a scaled experimental technique to measure the stability of individual structures and folds in a protein. These individual flexibilities within a protein are highly therapeutically relevant, implicated in many dynamic processes like amyloid formation or aggregation of therapeutic antibodies. His lab’s approach leverages hydrogen exchange (HdX): when proteins in water are diluted into D2O, unfolded polar hydrogens are replaced with deuterium while backbone hydrogens in stable secondary structures retain their bonds. As regions unfold, they undergo rapid deuteration, with their exchange rates denoting local stability.
By combining oligo pool libraries with LC-MS, his lab quantified how many residues in each protein exchanged quickly versus slowly across different D2O exposure times, measuring this distribution across 6,000 proteins. This allowed them to quantify cooperativity — how many residues only unfold together during complete unfolding. Surprisingly, when attempting to predict cooperativity from this dataset, they found their linear models with a small set of curated features outperformed language models. They additionally were able to train a model to predict plausible sequence variants to improve cooperativity, though that is still early. These experimental measurements provide crucial constraints for future dynamical models and MD simulations.
Erika DeBenedictis closed with a vision for large-scale public projects to advance predictive models for biology. Drawing from her physics background, she highlighted how biology has seen few coordinated scientific efforts in comparison to physics (i.e CERN). Even the PDB, while a transformative resource, emerged somewhat accidentally over 50 years at a cost of ~$10B.
Through her non-profit Align to Innovate, she argued we can dramatically accelerate this timeline by creating clear roadmaps, partnerships, repositories, and benchmarks for specific datasets and models. Their first goal is to tackle protein function with the goal of collecting data and training models that can transfer learnings between functional families (technical roadmap). They propose using GROQ-Seq, a high-throughput, extensible assay that links cell growth to functional outputs to aggregate this dataset.

Success will require broad stakeholder buy-in and Align has already garnered support across academia, industry, and government bodies like NIST. It felt especially fitting that she concluded her talk by turning to the audience, polling them on what techniques they would like to see go head-to-head (somehow sequence vs structure edged out David Baker vs AlphaFold) or what functional data they preferred (antibodies won above transcription factors).
While parts of the field feel mature - AF2's release seems distant now - we're likely still in the early innings of its journey. These initiatives and projects could generate several PDB-scale datasets, enabling more Nobel-worthy breakthroughs. At Dimension, we're excited to witness this era of rapid progress — please reach out if you're working on something here!
GenAI for Health
Workshop Summary
“Generative AI (GenAI) emerged as a strong tool that can revolutionize healthcare and medicine. Yet the public trust in using GenAI for health is not well established due to its potential vulnerabilities and insufficient compliance with health policies. The workshop aims to gather machine learning researchers and healthcare/medicine experts from both academia and industry to explore the transformative potential of GenAI for health. We will delve into the trustworthiness risks and mitigation of cutting-edge GenAI technologies applicable in health applications, such as Large Language Models, and multi-modality large models. By fostering multidisciplinary communication with experts in government policies, this workshop seeks to advance the integration of GenAI in healthcare, ensuring safe, effective, ethical, and policy-compliant deployment to enhance patient outcomes and clinical research.” —Workshop Website
Speaker Summaries
Improve Medical GenAI Model’s Trustworthiness with Expert Knowledge and Synthetic Data (Daguang Xu, NVIDIA)
Dr. Xu began his presentation by stating that model trustworthiness is a roadblock for the clinical adoption of generating modeling techniques.
Gaps in model trustworthiness stem from issues with hallucination, safety, bias, and privacy.
Hallucination is a tendency of most generative models wherein they produce ‘real-looking’, but false data that can be challenging to root out given the black-box nature of these algorithms. Model explainability is critical for adoption.
Safety issues occur more at the user-level. That is, if physicians don’t know how to audit model results prior to acting on them—mistakes can be made in a clinical setting. Xu mentioned two relevant efforts to help here: Llama Guard and Nemo-Guardrails.
Bias stems from inadequate training data. Diseases or patient groups underrepresented in the training corpus have worse performance.
Privacy is always a critical issue when handling patient data.
Dr. Xu focused his talk on hallucination and bias. Specifically, he argued injecting expert knowledge and synthetic data into GenAI models could remedy hallucination and bias, respectively.
First, we went over Project MONAI (Medical Open Network for AI)—a collaborative initiative between NVIDIA and several academic institutions.
We focused specifically on a multi-modal model (VILA-M3) that serves as a VLM—a visual language model. It’s trained to input and reason over text as well as images (e.g., from medical scans).
M3 is built on the idea that general-purpose models are too prone to make errors, especially when given very specific data types or tasks. To mitigate this, VILA-M3 triggers specific, expert workflows from other models contained within Model Zoo.
For example, when prompted with a thoracic CT image, VILA-M3 launches the expert model VISTA3D which can segment and analyze these types of images.
Second, we dealt with issues relating to model bias. This issue emanates from the long-tail problem in healthcare, that is when certain diseases are underrepresented in the training corpus and thus less likely to be classified correctly.
Dr. Xu postulated that augmenting datasets with synthetic data can increase performance in these cases.
Responsible Radiology Reporting in the Age of GenAI (Tanveer Syeda-Mahmood, IBM, Stanford)
In this presentation, we again focused on medical imaging applications of VLMs in a radiographic setting. Specifically, Dr. Syeda-Mahmood discussed using VLMs to create generative radiology reports prior to manual review by radiologists.
Responsible radiology reporting is not just about being accurate about what pathologies might be in an image—but also specific about what’s not found in an image. Models should avoid irrelevant descriptions—they shouldn’t be too assumptive or inclusive of information not contained in an image.
Recall that many generative models were originally designed for creativity. If one prompts a radiographic model like Xray-GPT, it will generate different reports each time, which creates a lack of standardization that’s problematic in a medical context.
Dr. Syeda-Mahmood was careful to mention methods like RGRG that give stable reports, though the responses are canned and variety is missing—causing a trade-off.
Responsible radiology reporting is critical because even if models are FDA cleared, they must earn clinician trust—an uphill battle. Models should be sensitive to demographic biases. They should be able to distinguish between an actual lack of evidence versus omission.
AI Agents for Biomedical Discoveries (James Zou, Stanford)
Agentic AI was a theme of this workshop—and indeed a theme of the whole conference. AI agents require thinking about AI as a partner rather than just a tool.
Professor Zou discussed creating a virtual lab in a multi-agent framework, where specialized agents communicate with each other to solve a problem.
These semi-autonomous assemblies of agents have group meetings wherein they debate how to approach complex tasks. In their work, the group emulated both group and individual (1:1) meetings between agents to design nanobodies against a SARS-CoV-2 variant. The bots decided which computational tools to use, wrote additional code, and nominated a host of sequences that were tested in a lab. Read more in The Virtual Lab.
Demographic Bias of Expert-Level Vision-Language Foundation Models in Medical Imaging (Yuzhe Yang, UCLA)
A major issue I’ve observed studying companies working in digital histopathology is the issue of generalization. Algorithms designed to make diagnoses or accelerate radiologist workflows sometimes see degraded performance in new health systems—whether that’s because of different instrumentation or changes in the underlying patient demographics.
Professor Yang’s talk focused on uncovering and auditing the biases of VLMs, which can lead to inadequate treatment in the real world, such as under-diagnosis in less well-represented patient cohorts. (Source)
The research team assembled chest x-ray images (n>800,000) from five diverse datasets (e.g., CheXpert). (Source)
Next, they tasked a host of contemporary PLMs to perform zero-shot diagnoses using these images. While many overall AUC values were on par with individual radiologists (>0.9), the group fairness scores suffered significantly. This means that the VLMs showed statistically significant underperformance relative to radiologists on underrepresented patient cohorts.
PATIENT-Ψ: Using Large Language Models to Simulate Patients for Training Mental Health Professionals (Stephanie Milani, Carnegie Mellon)
Stephanie Milani delivered an outstanding presentation on addressing mental healthcare gaps using LLMs.
The World Health Organization (WHO) notes that 1-in-8 adults suffer from a mental health condition. Over half of US adults aren’t receiving mental healthcare services, partially due to inadequate therapist supply. Stephanie’s presentation dealt with the extent to which technology can mitigate this burden.
After interviewing many mental healthcare professionals who felt their training inadequately prepared them to address real patients, the research group asked whether LLMs could be leveraged to simulate real-world patients, thereby improving provider readiness.
Models like Chat-GPT cannot simulate patients out-of-the-box. Instead, Patient-Ψ seeks to first build cognitive models that mirror the typical behavior, emotions, thought patterns, and pathologies of patients with real psychological disorders. Constructing a patient’s cognitive model is a key aspect of cognitive behavioral therapy (CBT).
Unfortunately, there are no open databases of cognitive models. The Patient-Ψ team utilized GPT-4 to create initial summaries of real therapy sessions. Real therapists then interacted with this formless summaries, generating 106 rich, diverse cognitive models using human expertise.
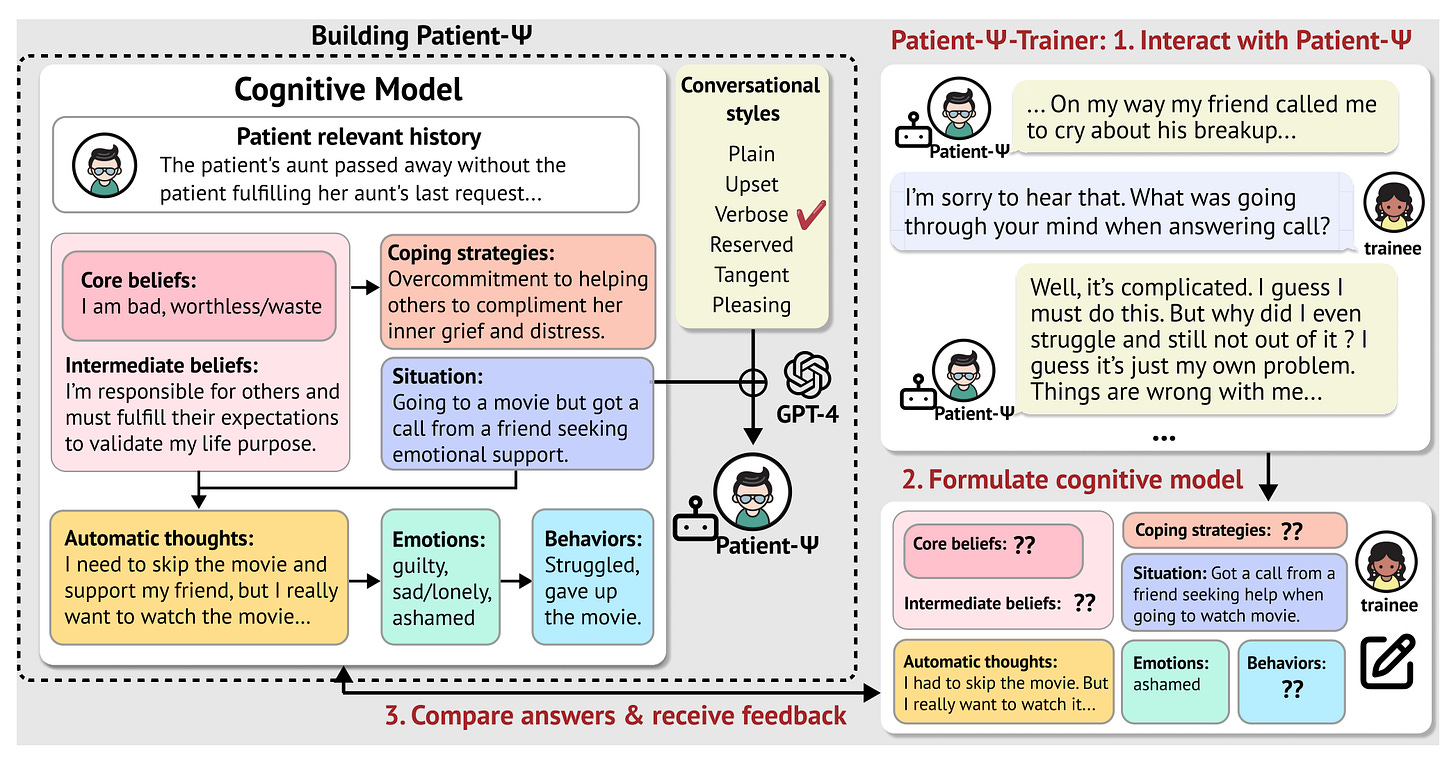
This diagram shows the procedure the team used to build Patient-Ψ, including expert feedback. (Source) My understanding is that the research group also used a conversational tone filter that augments and injects complexity into the cognitive models during inference time to better prepare trainees.
Using 20 professionals and 13 trainees, the researchers evaluated Patient-Ψ against GPT-4 to assess whether the former more closely resembled bonafide cognitive patient models and whether it improved trainee readiness. The following plot summarizes the results:


Position: Participatory Assessment of Large Language Model Applications in an Academic Medical Center (Giorgia Carra, CHUV; Lausanne University Hospital)
Dr. Carra brought a unique perspective as a senior data scientist employed at the University Hospital of Lausanne in Switzerland. Her work involves improving care quality and operational efficiency using ML techniques.
Manifesting this vision involved assembling a hospital working group composed of 30 diverse healthcare professionals. These experts participated in moderated, multi-round discussion sessions to first identify and then score LLM use cases within the context of their healthcare system. The group’s output is shown below.

These scores reflect the working group’s output across multiple conversations. Lower numbers are better. (Source) In the eyes of regulators—LLMs are not technologies unto themselves. Instead, the United States FDA, European Law on Medical Device Regulations (MDR), and the European Union (EU) AI Act apply varying regulatory frameworks depending on the intended LLM use case. (Source)

Based on their own experience and the most recent stipulations of the aforementioned regulatory frameworks, the working group voted automatic incident report classification and adaption of medical documentation into patient-amenable language as the two use cases most likely to be safe, compliant, feasible, and impactful.
It will be interesting to follow this group’s work as they execute on this action plan and learn more about the deployment of healthcare LLMs into a prospective hospital setting.
Generative AI for Next-Generation Healthcare: Where Are We Now? (Yuyin Zhou, UC Santa Cruz)
We began by reviewing several hot uses cases for GenAI in healthcare, including alleviating administrative burdens via LLMs, assisting with differential diagnoses using imaging data, supporting medical decision-making broadly, and predicting disease risk.
Like other presenters, Professor Zhou remarked that data shortages may arrest the field’s rate of progress. Moreover, she noted that the lack of multi-modal data alignment is a challenge. This means that not all data across modalities is well annotated. For example, radiographic images might be lacking defined regions of interest (ROIs) or detailed diagnostic descriptions useful for model training.
MedTrinity-25M is a large, semi-synthetic, multi-modal dataset spanning 25 million images with ROI and text annotations across 10 imaging modalities, 65 kinds of diseases, and 48 kinds of anatomical structures. As shown below, the authors leveraged a multi-modal LLM to enrich the original data with detailed annotations. (Source)

MedTrinity-25M constitutes a rising tide—that is, pre-training using MedTrinity-25M boosts the performance of other vision-language models (VLMs).
Critically, Professor Zhou highlighted several shortcomings of this approach, including inaccurate descriptions, non-specific disease annotations, ambiguous local-global relationships, and more. The talk concluded with the mention of several newer evaluation benchmarks that more closely track clinical utility, such as LancetQA and NEJMQA. (Source 1, Source 2)

Risk Assessment, Safety Enhancement, and Guardrails for Generative Models for Health (Bo Li, U. Chicago, Virtue AI)
GenAI model safety and trustworthiness are key concerns amongst healthcare stakeholders.
Professor Li walked through several examples of LLM jailbreaks—techniques that can be used during inference time to trick a model into violating its safety programming. For example, users can force a specific output format (e.g., YAML) or ask the model to tell a “fictional” story that contains instructions on how to make a salad with a poisonous mushroom.
I appreciated that we walked through open-source LLM safety and regulatory compliance leaderboards (e.g., AirBench). These include benchmarks for performance, evaluation reports, and red-teaming strategies to make models robust against tampering. (Source 1, Source 2)
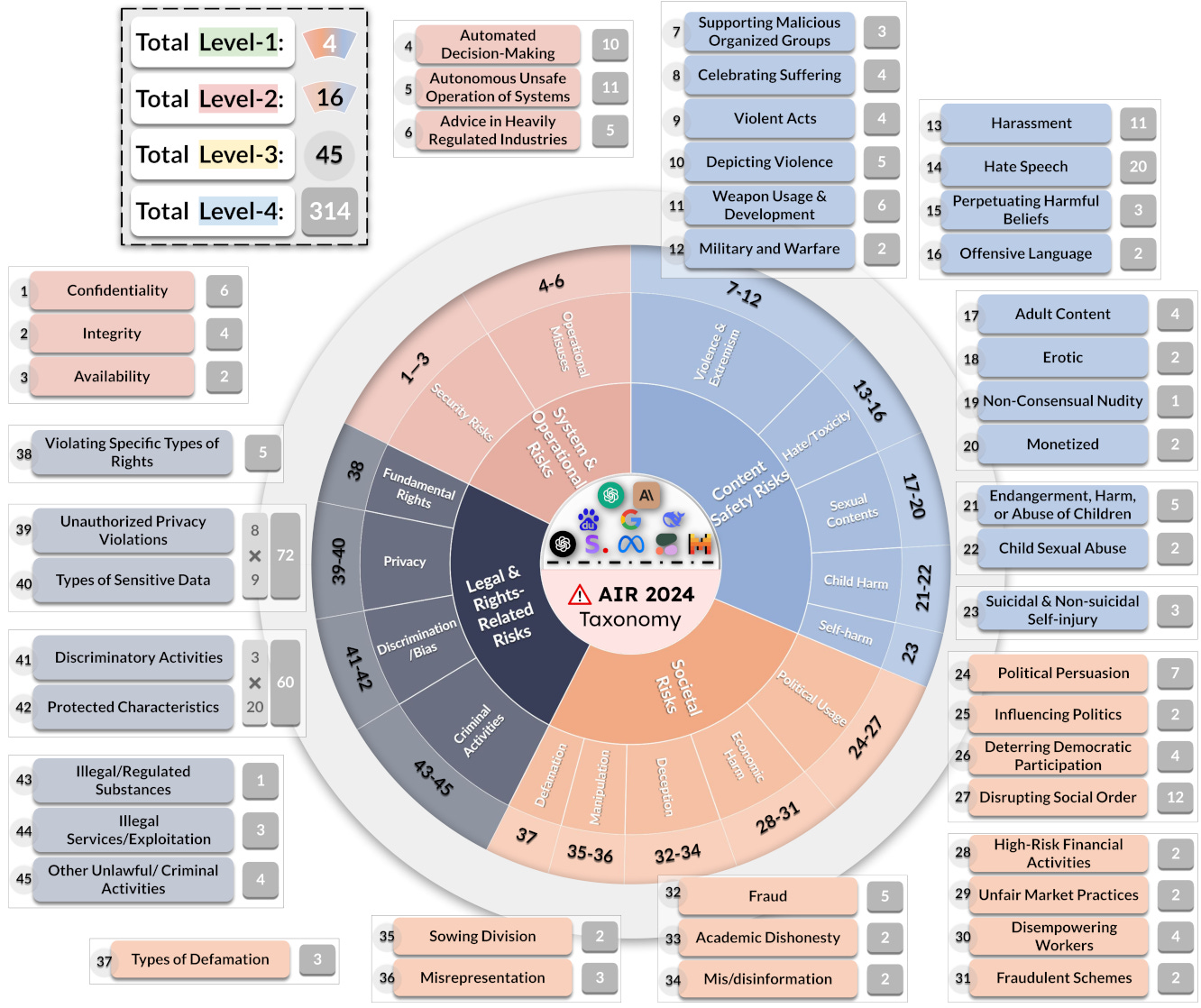
AirBench is the first LLM safety benchmark aligned with emerging government regulations and company policies. It includes a framework and taxonomy for evaluating potential safety liabilities. (Source) Professor Li concluded by discussing GuardAgent, her group’s effort to mitigate safety and compliance obstacles with medical LLMs. GuardAgent is an LLM agent that inputs user-defined safety settings and analyzes the output of an upstream generative model. Should GuardAgent flag any safety violations, it can filter or deny the original output. (Source)
Context Clues: Evaluating Long Context Models for Clinical Prediction Tasks on EHR Data (Sanmi Koyejo, Stanford)
We focused on electronic health record (EHR) foundation models. Typically, these LLMs are trained on large bodies of EHR data to power downstream tasks like generating a medical discharge summary or making predictions about a patient’s future health status. (Source)
EHRs are complicated data objects—quite unlike standard written text. A patient’s EHR is a timeline of clinical events split into multiple parallel tracks including diagnoses, prescriptions, lab tests, procedures, healthcare visits, and more.
It’s standard practice to compress EHR data into a single, tokenized vector before passing the tokens through a sequence model (e.g., GPT) for generative tasks.
Many contemporary LLMs have relatively short context windows. This challenges the use of EHR foundation models given lengthy patient histories.
Given the underlying nuances and extended duration of EHR data, this talk focused on two guiding questions:
How to the unique properties of EHR data affect LLM performance?
Does long context outperform short context for EHR data?
The researchers tested four architectures (GPT, Llama, Hyena, and Mamba (~120M parameters)) across context lengths ranging from 512 to 16k. They tested performance against the EHRSHOT benchmark for few-shot evaluation of EHR models. (Source)
Perhaps unsurprisingly, the longest context (16k) Mamba model was most performant based on mean AUROC across 14/15 EHRSHOT tasks.
This experiment highlighted three, specific EHR data nuances that all detract from LLM performance.
EHR data is often used for medical billing and coding. Therefore, the data contains repetitive strings of billing/coding text that degrade LLM performance.
Clinical events have irregular time intervals—some interactions take minutes while others can take years. Irregular token intervals also create performance issues.
Unlike natural text, the perplexity of EHR tokens increases over time. Consider that in standard sentences, the last word is usually the easiest to guess. If I say, “Eeny, meeny, miny, ___!”—you can probably guess the last word in the phrase. EHR data is the opposite. The longer the clinical histories are, the more complex and hard to model the morbidities might be, leading to increasing uncertainty in predicting next tokens.
It’s clear the field needs to experiment with longer context models as well as develop special handling instructions to contend with the idiosyncrasies of EHR data.

AI for Novel Drug Modalities
Workshop Summary
“The primary objective of this workshop is to bridge the gap between AI and emerging drug modalities, such as gene and cell therapies, and RNA-based drugs. These modalities are important for disease mechanisms which were previously considered difficult, if not impossible, to target. They offer novel mechanisms of action, target previously undruggable proteins, and can address unmet medical needs with higher precision and efficacy. Traditional modalities such as small-molecule drugs, recombinant proteins, and monoclonal antibodies have formed the basis of numerous existing treatments. Recently, there has been growing recognition that AI methods have the potential to expedite breakthroughs in these established modalities. This same potential extends to the emerging field of new drug modalities, where AI can play a transformative role in accelerating the discovery and development process, overcoming challenges, and ultimately bringing innovative therapies to patients more efficiently and effectively. We aim to bring specialists in these modalities and ML communities together to discuss and investigate how AI can accelerate drug development in these emerging modalities.” —Workshop Website
Machine Learning-Guided Sequence Design for mRNA and Gene Therapy; Georg Seelig (University of Washington)
Gene therapies acting on DNA and RNA hold significant promise for treating diseases linked to aberrant gene expression.
Despite this promise, a largely unsolved roadblock in gene therapy has been the ability to control both gene expression magnitude and its specificity to select cell types in vivo.
Prof. Seelig and his group studied and designed synthetic regulatory elements that can control these two aspects of gene expression (i.e. magnitude and specificity).
Regulatory gene elements are a class of DNA sequences that influence when, where, and how much a gene is expressed.
Enhancers are one type of regulatory gene element defined as non-coding sequences of DNA that contain transcription factor binding sites (TFBS) that, when bound, can directly regulate downstream gene expression in a cell-type specific manner.
These enhancers can range from 100s-1000s of base pairs in length, contain several other sequence elements aside from TFBS, and are often distally located from the genes they act upon, making it difficult to understand and design novel enhancers that are unique to specific genes and cell types of interest.
To solve this problem, the authors leveraged a deep-learning framework to identify novel cell-specific enhancers by iteratively training their model with two complimentary datasets: (1) chromatin accessibility and (2) gene expression collected from massively parallel reporter assays (MPRAs).
Chromatin accessibility:
Open chromatin is often associated with enhancer regions because transcription factors need to physically access and bind the DNA to induce downstream gene expression.
The authors used accessibility data collected by mapping DNase I hypersensitivity sites (DHSs) (>750k measurements). DNA that is less densely packed (i.e. open chromatin) is more physically accessible to the DNase enzyme and therefore susceptible to cleavage. Subsequently, cleaved DNA fragments are read out through high-throughput sequencing techniques for quantification of open chromatin sites.
However, solely relying on chromatin accessibility data to identify enhancer sequences is limiting - just because a region of DNA is theoretically “open”, does not mean that the sequence is an enhancer or that a transcription factor will be able to selectively bind to it. Hence, the need for the next dataset.
Massively parallel reporter assays (MPRAs):
MPRA is a high-throughput method to screen regulatory elements in vitro through the introduction of plasmids containing unique enhancer sequences upstream of a unique genetically-encoded barcode. Quantification of enhancer activity for each plasmid is measured via high throughput sequencing by comparing the ratio of plasmid DNA barcodes to RNA barcodes expressed with the reporter gene.
The team utilized an MPRA dataset containing 30k measurements to train a convolutional neural net for predicting cell-specific enhancers for either K562 or HepG2 cell lines, enabling them to identify synthetic enhancers that outperform natural genomic sequences.
However, MPRA data is limited by the cell lines used to collect the data, and may not be representative when trying to apply the learnings from one cell type to another.
Given the unique advantages and limitations with both of these datasets, the authors leveraged the experimental validation data from each model above as inputs to train a unified multimodal model.
Compared to training on individual data types, with this approach, they saw significant improvements in their model’s performance resulting in identification of enhancers that were ~100x more specific to HepG2 cells than K562.

An outline of multi-round modeling of both MPRA functional screening data (left) and DNA accessibility data (right), which was subsequently combined into a single multimodal model in “Round 2” (bottom). (Source)
Future work from the group is also focused on training deep learning models from large accessibility datasets collected from 733 human samples encompassing 438 cell and tissue types. With this model they aim to predict novel enhancers across many cell types and validate them in vitro using MPRA experiments.
Interested in learning more about this work? Check out the authors related preprint on bioRxiv!

Old Friends & New Partners: Accelerating Genome Engineering with Fine-Tuned LLMs and CRISPR-GPT Agent; Le Cong (Stanford)
CRISPR-Cas9 gene editing (and its many derivatives) has truly revolutionized our toolkit for understanding and engineering the human genome, and Prof. Cong has been right at the forefront of this field.
The Cong Lab has positioned themselves at the intersection of both the wet and dry lab when it comes to leveraging CRISPR. Their previous work has explored (1) the use of CRISPR for high-throughput genome engineering, (2) the development of single-cell technologies like Perturb-Track to study immune diseases, and (3) the construction of novel AI/ML tools for democratizing gene editing.
Prof. Cong’s talk at this workshop focused on this third pillar, specifically presenting on how AI agents and biological foundation models can be leveraged to perform complex tasks that will accelerate our adoption and advancement of CRISPR-based tools.
He touched on the idea that existing general purpose LLMs (e.g. ChatGPT) are currently not well suited to excel at complex tasks in biology. For example, even GPT4 models only score ~60% when asked PhD-level scientific questions from the GPQA Diamond test, and they cannot design novel guide RNAs for a CRISPR target when prompted.
Instead, field-specific AI agents (the “new partners” he is referring to in the title of his talk) trained on biological datasets should take place as the new gold standard for guiding biologists in experimental design and analysis.
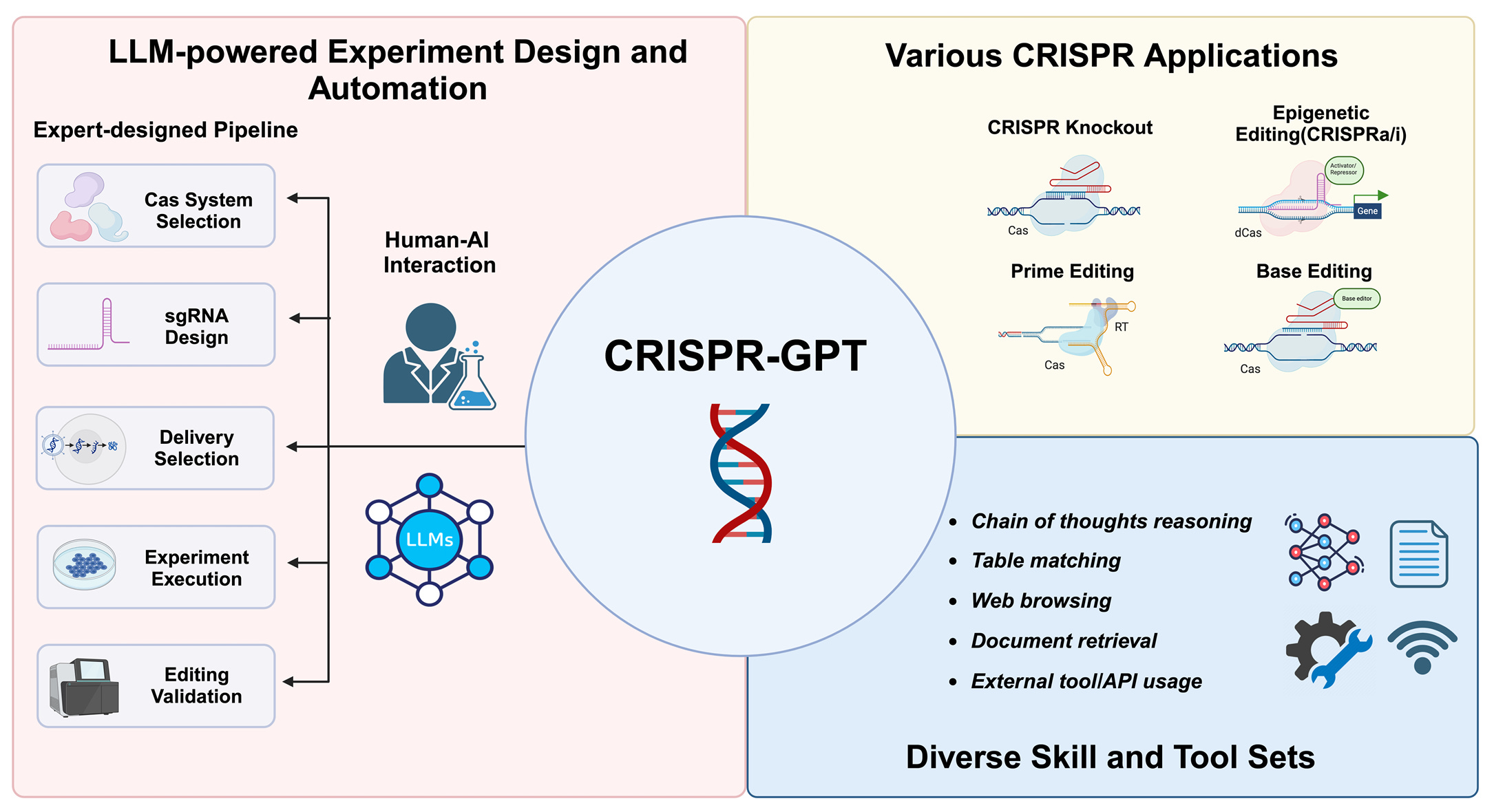
To this end, they introduce CRISPR-GPT, a purpose-built AI agent for genome/cell engineering, with three distinct “modes” that dictate its output: (1) Meta Mode, (2) Auto Mode, and (3) Q&A Mode.
For all the following modes, CRISPR-GPT leverages advanced LLM capabilities (e.g. Chain of thought, instruction fine-tuning), computational skills (e.g. web searching, code generation), and bioinformatic tools (e.g. Primer3, CRISPRitz) to execute on each request by the researcher.\

Overview of CRISPR-GPT capabilities and toolsets. (Source) In Meta Mode, researchers can get guidance on gene editing workflows that are pre-defined by experts in the field. For example, one could select a task from a curated list such as “Generate a knockout using CRISPR for Gene X.” The agent would then take you through a step-by-step guide on selection of the sgRNA, experimental conditions, analysis techniques, and more.
In Auto Mode, researchers can prompt CRISPR-GPT in natural language to receive customized guidance on their own workflows. For example, one can ask “Which CRISPR system should I use to repress TGFBR1 expression and how should I design my sgRNA?” CRISPR-GPT will then create an on-demand guide to inform and execute their experimental design.
In Q&A Mode, researchers are free to ask CRISPR-GPT ad-hoc questions related to field norms, experimental design, method comparisons, and much more. For example, one can ask “What are the differences between Cas12a and Cas12f?” and get back real-time answers to directly inform research directions.
Grounded in language models like ChatGPT, tools like CRISPR-GPT have significant potential to redefine the way we design and run experiments. Using the platform, they showed that CRISPR-GPT running in Auto Mode on top of GPT4o doubled accuracy scores in their benchmarking tests compared to GPT4o alone. Benchmarking included tests on experimental design, sgRNA design, delivery selection, CRISPR knowledge, and more.
Interested in learning more about CRISPR-GPT? Check out the preprint on arXiv!
Toward AI-Driven Digital Organism: Multiscale Foundation Models for Predicting, Simulating, and Programming Biology at All Levels; Eric Xing (GenBio AI, CMU, MBZUAI)
Modeling biology is inherently a multi-scale endeavor, from quantum interactions between atoms in a protein to the cross-talk of different organ systems within a patient.
To start the talk, Dr. Xing proposed an exciting question: “How do we combine all of these length scales into a single foundation model?”
Notably, in Dr. Xing’s view, biological education and practice has traditionally been siloed between “departments” (e.g. biochemistry, molecular biology, cell biology, tissue biology, systems biology, etc.) as well as a delineation between organ systems (cardiovascular, neural, gastrointestinal, etc.).
The ability to develop novel treatments for diseases often requires our unified understanding of many different scales of biology at once, and this current organizational framework limits interdisciplinary innovation and the ability to holistically model biological systems.
Biology isn’t in a vacuum. It always has a context. How do we bring this context into play?
To answer this question, Dr. Xing and the team at GenBio proposed a vision towards large foundation models (FMs) that can help us holistically understand and engineer biology across many different length scales. In this talk, he outlined a roadmap for building out a unified and integrated system of FMs for biology, which he broke down into three stages: (1) Divide and Conquer, (2) Connect the Dots, and (3) Align and Optimize Across Scales.

GenBio’s proposal for an inter-connected system of foundation models, termed an AI-driven digital organism (AIDO), that leverages both public and proprietary biological datasets. (Source) In Divide and Conquer, they proposed a build-out of several FMs unique to specific biological scales and functions. These FMs include individual sequence and structure FMs for the molecular scale, DNA, RNA, and proteins, as well as an Interactome FM, Cell FM, Tissue FM, and Phenotype FM. These FMs are trained from integrated public and proprietary datasets (e.g. Genebank, Uniprot, KEGG, ENCODE, TCGA, Human Protein Atlas, etc.).
In Connect the Dots, they proposed a framework to integrate several models together to better inform certain biological tasks that require a richer representation of the surrounding environment. Notably, Dr. Xing emphasized the need for integration of both sequence- and structure-based FMs for tasks such as the inverse folding problem to predict sequence from structure.
In Align and Optimize Across Scales, they proposed co-tuning these models to work more effectively together at more complex modeling tasks. Applications of a fine-tuned, holistic FM may include applications in cell engineering such as predicting the effects of drug or gene knockout perturbations or applications in phenotype engineering such as predicting how genetic mutations could impact disease progression and inform dietary suggestions.
Following this walkthrough, the talk ended with a call for re-defining the way we view the proverbial “lab-in-a-loop.” The wet-lab of the future should not be considered a search tool, but instead a tool for validation. The computer should take its place as the search tool.
Want to learn more about their proposal for the future of biological foundation models? Check out their preprint on arXiv!

DeepADAR: Modeling Regulatory Features of ADAR-Based RNA Editing for gRNA Design; Andrew Jung (Deep Genomics, U. Toronto Vector Institute)
Adenosine deaminase acting on RNA (ADAR) based editing platforms are garnering a growing interest among researchers for their ability to both (1) correct genetic mutations on the RNA level and (2) be used as a broader platform for controlling gene expression.
ADAR editing platforms work by making an A-to-G edit on double-stranded RNA (dsRNA) molecules.
Therefore, in order to target a specific RNA of interest in the cell, one must design and deliver a guide RNA (gRNA) that forms a dsRNA complex complimentary to the target of interest.
Two key interrelated determinants for whether the ADAR enzyme successfully edits the target sequence are (1) the presence of specific base mismatches along the dsRNA complex and (2) its underlying 3D structure.
This poses a significant challenge for researchers wanting to leverage ADAR against a specific RNA target: the underlying rules that dictate where these mismatches should be placed and the requirements for a proper 3D structure are not well defined, and the design space is often too large for experimental methods to be exhaustive.
To address this problem, Andrew Jung and his colleagues trained a deep learning model to learn regulatory features of RNA editing and predict gRNA editing against a particular target of interest.
Current databases of ADAR editing sites are limited and contain a substantial number of false positives. These datasets can be filtered for higher quality editing sites through the use of public whole genome sequencing (WGS) and RNA-seq datasets that inherently provide greater evidence of RNA editing by ADAR.
This information is then used for model training: for a given region of RNA, output a scalar value for every “A” in the RNA sequence predicting the probability of editing.
When running the model, the input is both RNA sequence and structure, as both of these characteristics are well-known to impact the efficiency of ADAR editing, especially in the context of predicting gRNA-induced editing.
At the end of the talk, they evaluated DeepADAR’s ability to predict both endogenous RNA editing and gRNA-based editing.
Endogenous RNA editing was predicted using a CRISPR mutagenesis dataset covering 366 mutations across 3 genes. Their results show an average predictive score across the 3 genes of ~0.85 (area under receiver operating characteristic curve (auROC)). For some context, a score of 0.5 is equivalent to random guessing, while a score of 1 is perfect prediction.
To predict gRNA-based RNA editing, the authors experimentally measured editing efficiencies of 240 gRNAs from ~80 unique editing sites across 6 genes. By fine-tuning the previous endogenous model on their experimental data, they were able to achieve a ~0.75 spearman correlation for on-target editing performance.
Future directions for the team involve making these models more tissue and cell-type specific (as endogenous ADAR expression and activity is known to be higher in certain cell types) and incorporating information on chemical modifications that impact RNA structure.
Adaptyv Bio Protein Design Competition; Julian Englert (Adaptyv Bio)
We also had an update from Julian Englert, Adaptyv Bio’s CEO, on their Protein Design Competition.
For the competition, they asked protein designers from around the world to submit designs for binders against EGFR.
EGFR plays an important signaling role in driving cell proliferation and differentiation across several tissue types.
Mutations in EGFR and mis-regulation of its expression have been associated with several diseases, most notably cancer.
Current FDA-approved mAb therapies targeting EGFR (e.g. Cetuximab) have demonstrated the promise of inhibiting EGFR transduction and paves the way for novel protein binders in this space.
For the competition, Adaptyv Bio received over 1000 protein designs sourced from 100 different designers.
These designs were ranked in silico based on: (1) computational metrics from AlphaFold, IPSA, ESM2, among others and (2) how unique and interesting the design approaches were.
The top candidates were selected and tested in Adaptyv Bio’s lab, to which there were ~50 validated binders out of ~400 total designs.
Two of the winning designs from (1) Aurelia Bustos and (2) Constance Ferragu were featured at the workshop.

Overview of protein designs from the top three winners of Adaptyv Bio’s Protein Design Competition. (Source) Their approach was to optimize natural EGFR ligands such as EGF, TGF-alpha, Betacellulin, and others, with the rationale that binders based on endogenous ligands with specific mutations could potentially be good candidates.
For each of the ligands, they computationally identified the probabilities of contact between each animo acid with EGFR using AF3 and kept these amino acid residues in their final design if they were conserved amongst 5 of the endogenous binders or if the contact probability was > 0.9.
They then masked any amino acids that did not satisfy these criteria and used ProteinMPNN to comprehensively explore variants of the masked regions using inverse folding (i.e. sequence prediction from structure), generating 1000s of novel amino acid sequences.
Finally, they trimmed these designs based on a ranking pipeline of computationally-predicted metrics and validated final structures using AlphaFold and Chai1.
Interestingly, they found that only those designs derived from EGF and Betacellulin had success in the lab.
Their top design resulted in a binding affinity of 51.6 nM, and you can check out more details of their that binder here!
Constance Ferragu (Cradle Bio)
“It took us 30 minutes over some kombucha.” This was the opening line from Constance when answering the question “What did it take?” to make their binder design.
More specifically during that 30 minute window, the team started with scFv-formatted Cetuximab as their base protein and constrained themselves (as per the competition’s rules) to impose a minimum of 10 mutations in their final construct.
Interestingly, they did not make any mutations within the complementarity-determining region (CDR) (i.e. the variable domains on the light and heavy chains that directly interact with the ligand of interest) of the original Cetuximab antibody. Instead, they focused on introducing mutations within the framework regions (i.e. the relatively conserved regions flanking the CDR) in a process referred to as framework engineering.
With those constraints defined, they performed an overnight run of the algorithms developed at Cradle, which resulted in a binder with 1.21 nM binding affinity against EGFR (8x greater than the Cetuximab scFV).
Check out the details of their top binder here!
Want to see more details on all of the contest entries? Check out the website here!
